1Hospital for Children and Adolescents, 2Department of Diagnostic Radiology, Paediatric Radiology, 3Institute of Laboratory Medicine, Clinical Chemistry and Molecular Diagnostics, University of Leipzig, Germany
Combined pituitary hormone deficiency (CPHD) is a rare disorder resulting from impaired production of several pituitary hormones. This report describes the 30-year history of a patient who has apparently lived without a pituitary. The patient, born in 1976, experienced recurrent episodes of hypoglycaemia (as low as 2.9 mg/dl) in early childhood and showed elevated liver enzymes up to the age of two years without specific diagnosis. At the age of approximately 13 years, he first presented at our Department of Paediatric Endocrinology (University Hospital for Children and Adolescents, Leipzig) with signs of hypopituitarism and a height SDS of – 5.4, a high pitched voice and hypogenitalism. Endocrine testing confirmed panhypopituitarism (GH 1.3 ng/mL, LH 0.8 mU/mL, FSH 0.1 mU/ml, ACTH <1.0 pmol/l, TSH 1.0 mU/l) and substitution therapy was initiated. Magnetic resonance imaging (MRI) of the sella turcica showed a planar, hypoplastic and empty sella with pituitary stalk aplasia and ectopic neurohypophysis. Currently nearly 31 years of age, though taking his medication irregularly or not at all, he claims to be in good mental and physical condition irrespective of compliance or non compliance with substitution therapy. This case illustrates the importance of continued follow-up in patients with hypopituitarism and a systemic transferral of adolescents with CPHD to the care of adult endocrinologists.
Empty sella, GH deficiency, Panhypopituitarism, Pituitary hormone deficiency, Pituitary stalk hypoplasia, Ectopic neurohypophysis
INTRODUCTION
Hypopituitarism was first described by Simmonds (1914) who defined it as the inability of the pituitary gland to provide the body with hormones of the hypothalamic-pituitary-axis, like adrenocorticotropic hormone (ACTH), prolactin (PRL), growth hormone (GH), thyroid–stimulating hormone (TSH) and gonadotropins adapted to the needs of the organism.1 Childhood hypopituitarism may be acquired or of congenital/genetic origin. Possible causes leading to acquired hypopituitarism include a number of inflammatory, granulomatous or neoplastic diseases as well as infections, vascular anomalies and traumatic or radiation injuries involving the hypothalamic-pituitary region.1-4 Affected patients typically present with growth failure during childhood or adolescence. Moreover, growth hormone deficiency (GHD) is associated with decreased bone mineral density, impaired physical activity and an increased cardiovascular risk in adulthood as manifested by elevated fasting and postprandial serum lipid levels and increased body fat.1,5–8 Several congenital/genetic defects may be associated with pituitary dysfunction, like craniofacial abnormalities, empty sella or other midline defects.1,3,6,9,10 Among these, mutations in the PROP1 gene (prophet of Pit–1) are the most frequent genetic defects leading to CPHD.2,6,11,12
The prevalence of hypopituitarism is not well established and is most probably underestimated, with a reported figure of 300-455 cases per 1 million per year.3 Clinical symptoms such as fatigue, hypotension, cold intolerance, growth retardation or impotence and infertility in GH and gonadotropin deficiency can be non-specific, their severity depending on the extent of hormone deficiency.1,3,9 Establishing the diagnosis of hypopituitarism requires measurement of basal and stimulated serum values of anterior pituitary hormones and their affected hormones. Magnetic resonance imaging (MRI) of the hypothalamic-pituitary region is strongly warranted and in cases of CPHD genetic testing is recommended.3,9 Once hypopituitarism has been diagnosed, adequate replacement of all lacking hormones is required and should be monitored at regular intervals to provide the best clinical outcome.1,9,12 Generally, hypopituitarism is a chronic and lifelong disorder, unless the underlying disorder can be treated with resultant recovery of pituitary function. Patients with hypopituitarism seem to exhibit increased mortality.1,13,14
We report the medical history of a nearly 31-year old patient who lives without any visible anterior pituitary gland. At present, he is on substitution therapy but frequently neglects therapy over long intervals, during which time he nonetheless claims to be in good physical and mental health.
CASE REPORT
The patient is the second child (he has one healthy elder sister) of healthy, non-consanguinous parents of German descent. His father’s height is 172 cm and his mother’s height is 169 cm. He was born fullterm with a birth weight of 4120 g (+1.40 SDS) and a birth length of 53 cm (+0.60 SDS). Pregnancy and birth history were reported uneventful with the exception of a cephalhematoma and prolonged jaundice. At the age of 12 months he had a height of 69 cm and growth failure was suspected for the first time. Moreover, he had experienced several episodes of hypoglycaemia (2.9 mg/dl). However, no specific diagnosis had been made. In the following years, the boy showed progressive growth failure and bone age retardation (Figure 1). Up until 1989, he was placed under medical treatment several times at different hospitals in former East Germany, and starting in 1982 he received different formulations of human extractive GH (Sotropin H, Grorm) produced in the German Democratic Republic. During this time, Laron’s syndrome (GH resistance) was ruled out by IGF-I stimulation test after GH injections. Unfortunately, the exact data from these tests are not available.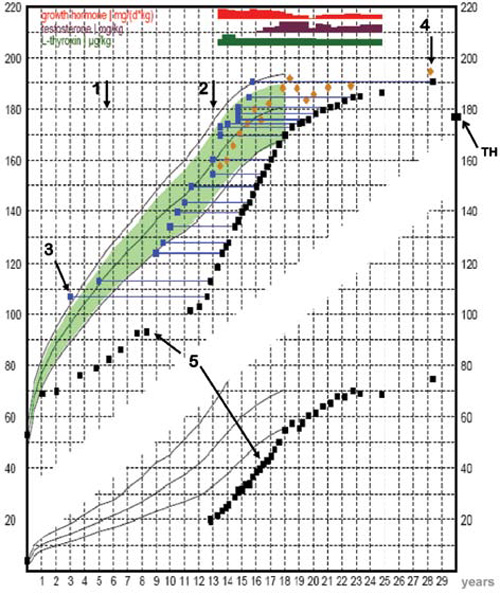
Figure 1. Growth chart of the patient with panhypopituitarism and empty sella. arrow 1 to arrow 2: intermittent administration of human extractive GH; arrow 2: start with recombinant human GH therapy. TH: target height; arrow 3: bone age; arrow 4: predictive adult height (according to Bayley and Pinneau); arrow 5: height and weight measurements.
Finally in October 1989, at the age of nearly 13 years, the boy was referred to the Paediatric Endocrinology Section of the University Hospital for Children and Adolescents, Leipzig. The first impression was that of overt panhypopituitarism. He was clearly too small for his age (Figure 2), his genitalia and gonads were small, he had cryptorchidism and had a somewhat older facial look with a high pitched prepubertal voice. There was a bone age retardation of 8 years (according to Greulich and Pyle) and the results of both basal and stimulated hormone levels (Table 1 ) confirmed the diagnosis of panhypopituitarism. In January 1990, and at a height of 114 cm (-5.32 SDS), therapy with recombinant human GH (1.5 IU/d) in combination with thyroid hormone (50 µg/d) treatment was started. Due to his still sufficient production of cortisol, no hydrocortisone substitution was necessary at this time. Under this treatment the boy showed a catch-up growth with an average growth velocity of 1 cm per month. In June 1992, he underwent an orchidopexy on both sides to correct his cryptorchidism and additional therapy with depot-testosterone (25 mg/month) was initiated. During the following two years, the dose of depot-testosterone was increased stepwise (100–125–200–250 mg/month). Despite this, he did not develop pubic hair and there was only a mild penile development to Tanner stage II. No significant increase of the plasma testosterone level could be observed. The subsequent intravenous pulsatile application of an LHRH analogue (100 µg Lutrelef) did not produce any LH, FSH surge. In April 1996, at the age of 194/12 years and with a height of 174 cm (-0.88 SDS), GH therapy was reduced to an adult supplementation. At this time, hypocortisolism with decreased ACTH levels was diagnosed and he was started on hydrocortisone. In the following years, he presented infrequently at our outpatient department. The attempt to transfer him from paediatric to adult care also failed due to his decreasing compliance. In November 2000 he stopped taking all his medications. At this time, he experienced such symptoms as pains in both knees, vertigo, fatigue and edemas when taking his GH and hydrocortisone. He repeatedly mentioned that he felt much better without medication. In 2001, he started training as a chef, working more than 12 hours a day without apparent problems. In his free time, he worked out or went cycling. He described himself as a loner with few personal friends and no sexual interests or contacts.
In November 2005, the patient presented again at our outpatient department. He had reached a height of 190.5 cm (+1.43 SDS) and was very pale. At that point, he presented the complete clinical and laboratory picture of non-treated panhypopituitarism. IGF–I was 12.85 µg/l (–10.6 SDS); IGFBP–3 was 0.87 ng/ml (–4.6 SDS); TSH 4.0 mU/l (reference range 0.4 – 3.5), FT4 6.7 pmol/l (10 – 25); LH <0.1 mU/ml (1.7 – 8.6); FSH 0.1 mU/ml (1.6 – 11.0); testosterone <0.07 nmol/l (9.9 – 27.8), and cortisol was 41.6 nmol/l (244 – 727 a.m.). He nevertheless felt generally comfortable except for the state of his genitals, which had remained at a stage of early puberty. Due to different changes in both job and life, he was planning to re-initiate treatment and in June 2007 he contacted our Paediatric Endocrinology Section again. He presented in a good general condition with a height of 192.3 cm (+1.69 SDS). IGF–I was 3.10 µg/l (–12.89 SDS); IGFBP–3 was 1.31 ng/ml (–3.52 SDS); TSH 3.30 mU/l, FT4 5.3 pmol/l; LH <0.1 mU/ml; FSH 0.1 mU/ml; PRL 68 mU/l (86 – 324), testosterone <0.07 nmol/l, estradiol 44 pmol/l (40–161), and cortisol was 17.3 nmol/l. He described himself as still resilient but also admitted taking hydrocortisone whenever he felt queasy. We commenced a new trial of therapy with 250 mg depot-testosterone every 4 weeks, 175 µg/d thyroid hormone and, as a starting dose, 10 mg per day hydrocortisone due to minor clinical signs of hypocortisolism but decreasing cortisol levels in laboratory testing.
Figure 2. Patient with panhypopituitarism and empty sella at the age of 1210/12 years; first presentation of the patient at the Paediatric Endocrinology Section at the University Hospital for Children and Adolescents, Leipzig, with an actual height of 112.5 cm (–5.4 SDS).
Since 1989, repeated laboratory investigations had showed normal levels of blood glucose, serum electrolytes (sodium, potassium, calcium) and creatinine. Serum lipids (cholesterol, triglycerides) were slightly increased, whereas LDL-cholesterol was still normal and HDL-cholesterol was slightly decreased. Hematological parameters (hemoglobin, hematocrit, erythrocytes) consistently showed levels slightly under the gender and age adjusted reference ranges. Serum alkaline phosphatase (AP) was consistently elevated (>6 µmol/ls; reference range <4.3) in contrast to liver enzymes that were intermittently elevated. The PRLvalues were normal or only slightly decreased. Dynamic tests at different ages are shown in Table 1 . Molecular genetic analyses of PROP1, LHX–3 and LHX–4 revealed no genetic defect.
Radiology: MRI of the sella turcica at the age of 233/12 years showed a planar, hypoplastic and empty sella filled with cerebrospinal fluid, a small bright zone of 1 mm near the optic chiasm which was suggestive of an ectopic posterior pituitary and no pituitary stalk (Figure 3). MRI of the hypothalamus and the supra- and infratentorial structures revealed no abnormalities, except a small structure at the left occipital pole suspicious for a small arachnoid cyst. Bone densitometry of the lumbar spine (L1 – L4) showed a z-score of –2.71. At the last presentation at nearly 31 years of age, bone age was more than 15 years with still open epiphysis.
Figure 3. MRI of the sella turcica at the age of 233/12 years, showing an empty sella (arrow), no pituitary stalk and a small contrast-medium accumulating zone near the chiasma opticum suggestive of a neurohypophysis remnant (discontinued arrow).
SUMMARY AND DISCUSSION
The patient herein reported was under medical treatment a number of times at different hospitals until panhypopituitarism was finally diagnosed rather late at the age of 131/12 years. We can only speculate that the anterior pituitary defect has existed since his first year of life as shown by his early predisposition to hypoglycaemia and his prolonged jaundice. It is speculated that trauma occurring at the time of delivery could have transsected the pituitary stalk, leading to a hypoplastic anterior pituitary with a progressive functional deterioration.4,14,16 Subsequently, regeneration of the distal axon of the hypothalamus could have formed a superiorly located posterior pituitary.4,14 In children with idiopathic GHD or CPHD, a higher frequency of breech delivery or podalic partus and of abnormal neonatal events has been reported. This could be a mechanism for stalk ischemia or trauma due to traction on the brain and spinal cord during delivery.4,14 In the paediatric literature, agenesis of the pituitary stalk is a frequently described syndrome.14,16,17 It is usually associated with CPHD or, more rarely, with isolated GH deficiency (GHD).4,17 Empty sella is reported to occur in between 5.5% and 23% of autopsy examinations of the sellar region.18 The exact pathogenesis of such MR abnormalities remains uncertain.4,14,16
However, the case herein reported is also noteworthy for other reasons. First of all, there is the patient’s particular growth curve (Figure 1), showing severe growth retardation during childhood and adolescence, followed by a spurt of growth under therapy with recombinant human GH (rhGH) under which he nearly reached his target height, followed in turn by a slow but continuous growth resulting in a height considerably taller than expected. Such tall stature in association with open epiphyses, distinct retardation in bone age and eunuchoid habitus is strongly reminiscent of patients with estrogen receptor mutations or aromatase deficiency. Hypogonadotropic hypogonadism with a prolonged lack of estrogen, estrogen insensitivity or aromatase deficiency results in eunuchoid skeletal proportions, reduced bone mineral density and open epiphyses.15,17,19–21 It is also well established that androgens do not effectively lead to epiphyseal closure without the combined action of estrogen. Thus, estrogen deficiency may be the major factor leading to retardation in bone maturation. However, in our case we can exclude estrogen deficiency as a possible factor for continuing growth due to low but normal estradiol levels probably resulting from adrenal and adipose tissue production. Estrogen receptor defects or estrogen insensitivity are also highly unlikely. It is speculated that factors other than GH or IGF–I could be important in determining height, e.g. insulin or PRL at high levels.15,17,19,22,23 But neither insulin nor PRL are elevated in our patient.
The second remarkable observation is the patient’s pubertal stage and genital development, which had remained at a stage of early puberty even though he had been instructed to undergo testosterone substitution. This observation is reminiscent of an androgen receptor defect or of partial androgen insensitivity syndrome (PAIS). However, the most likely explanation of failure of pubertal development in this patient was non-compliance. On repeated questioning, he admitted that he had received depot-testosterone injections only when hospitalised and did not allow injections at home. This accounts for the lack of plasma testosterone even “after several months of therapy” and his failure to advance during puberty. It is remarkable that, despite the lack of all anterior pituitary hormones and no or insufficient substitution therapy, the patient has evidently led an almost normal life without personal complaints except for lack of sexual interest. He has, however, admitted to taking hydrocortisone whenever he felt queasy. This means that he knew exactly how to overcome critical situations without taking medication regularly. Nevertheless, his cortisol levels have been consistently under the gender- and age-related norm during the last 10 years. This raises intriguing questions as to the manner in which he has been able to overcome stress and infections and to work up to 14 hours a day without sufficient hormone replacement. A fourth interesting point is the late manifestation of ACTH/cortisol deficiency. In patients with CPHD, GHD typically manifests early in life, whereas further hormone deficiencies, for unknown reasons, present somewhat later.9-11,13,24,25 Until the age of 19 years, his cortisol production seems to have been sufficient for him to lead an almost normal life. The delayed development of ACTH insufficiency could be due to progressive corticotroph apoptosis and secondary decompensation.6,11 A possible reason for this could be the lack of important paracrine signals from other pituitary cells inducing progressive cell death or apoptosis of corticotroph cells as has been suggested for patients with PROP1 gene defects.6
In summary, this case shows the natural history of an interruptedly treated panhypopituitarism due to agenesis of the neural pituitary stalk and once more highlights the fact that continuing growth can occur even in a patient with GH deficiency. Furthermore, this report illustrates the importance of long-term follow-up in patients with hypopituitarism and the necessity of establishing transition between paediatric and adult care.
ACKNOWLEDGEMENT
This work was kindly supported by CrescNet gGmbh, Leipzig.
SP and WK are supported by Deutsche Forschungsgemeinschaft, Bonn, Germany, DFG KFO 152 “Atherobesity”, project BE 1264/10-1 (TP 5).
REFERENCES
1. Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I, Stalla GK, Ghigo E, 2007 Hypopituitarism. Lancet 369: 1461-1470.
2. Vieira TC, Dias da Silva MR, Cerutti JM, et al, 2003 Familial combined pituitary hormone deficiency due to a novel mutation R99Q in the hot spot region of Prophet of Pit–1 presenting as constitutional growth delay. J Clin Endocrinol Metab 88: 38-44.
3. Ascoli P, Cavagnini F, 2006 Hypopituitarism. Pituitary 9: 335-342.
4. Hamilton J, Blaser S, Daneman D, 1998 MR imaging in idiopathic growth hormone deficiency. AJNR Am J Neuroradiol 19: 1609-1615.
5. Hull KL, Harvey S, 2003 Growth hormone therapy and Quality of Life: possibilities, pitfalls and mechanisms. J Endocrinol 179: 311-333.
6. Böttner A, Keller E, Kratzsch J, et al, 2004 PROP1 mutations cause progressive deterioration of anterior pituitary function including adrenal insufficiency: a longitudinal analysis. J Clin Endocrinol Metab 89: 5256-5265.
7. Geffner ME, 2003 Transition to the adult endocrine clinic: testing pituitary function – what tests and when? Growth Horm IGF Res 13: 117-121.
8. Karges B, Pfäffle R, Boehm BO, Karges W, 2004 Acromegaly induced by growth hormone replacement therapy. Horm Res 61: 165-169.
9. Geffner ME, 2002 Hypopituitarism and childhood. Cancer Control 9: 212-222.
10. Pfäffle R, Blum WF, 2000 Understanding the genetic of growth hormone deficiency. A reference guide. ISBN 1851132643. TMG Healthcare Communications Ltd.; UK.
11. Pavel ME, Hensen J, Pfäffle R, Hahn EG, Dorr HG, 2003 Long-term follow-up of childhood-onset hypopituitarism in patients with the PROP-1 gene mutation. Horm Res 60: 168-173.
12. Voutetakis A, Argyropoulou M, Sertedaki A, et al, 2004 Pituitary magnetic resonance imaging in 15 patients with Prop1 gene mutations: pituitary enlargement may originate from the intermediate lobe. J Clin Endocrinol Metab 89: 2200-2206.
13. Stanhope R, 2004 Transition from paediatric to adult endocrinology: hypopituitarism. Growth Horm IGF Res 14: 185-188.
14. Pentimone F, Riccioni S, Del Corso L, 1999 Congenital hypopituitarism in a 48-year old adult. Natural course, hormonal study and MRI evidence. Minerva Endocrinol 24: 87-90.
15. Wada S, Minagawa A, Imamaki K, et al, 2000 A patient of hypogonadotropic hypogonadism accompanied by growth hormone deficiency and decreased bone mineral density who attained normal growth. Intern Med 39: 641-645.
16. Navarro P, Halperin I, Rodriguez C, Gonzalez JM, Vidal J, Vilardell E, 1994 Congenital panhypopituitarism of late onset. J Endocrinol Invest 17: 347-350.
17. Ouden TT, Kroon M, Hoogland PH, Geelhoed–Duijvestijn PHLM, Wit JM, 2002 A 43-year-old male with untreated panhypopituitarism due to absence of the pituitary stalk: from dwarf to giant. J Clin Endocrinol Metab 87: 5430-5434.
18. De Marinis L, Bonadonna S, Bianchi A, Maira G, Giustina A, 2005 Extensive clinical experience: Primary empty sells. J Clin Endocrinol Metab 90: 5471-5477.
19. Fustini–Faustini M, Balestriere A, Rochira V, Carani C, 2003 Letter to the editor: The apparent paradox of tall stature with hypopituitarism: New insights from an old story. J Clin Endocrinol Metab 88: 4001-4004.
20. Smith EP, Korach KS, 1996 Oestrogen receptor deficiency: consequences for growth. Acta Paediatr: Suppl: 417: 39-43.
21. Smith EP, Boyd J, Frank GR, et al, 1994 Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331: 1056-1061. Erratum in: N Engl J Med (1995): 332: 131.
22. Tolis G, Cruess S, Goldstein M, Friesen HG, Rochefort JG, 1974 Survival and growth in a woman with untreated hypothalamic panhypopituitarism of 21 years’ duration. Can Med Assoc J 111: 553-556.
23. Kageyama K, Watanobe H, Nasushita R, Nishie M, Horiba N, Suda T, 1998 A hypopituitarism patient who attained tall stature without growth hormone. Intern Med 37: 472-475.
24. Cameron FJ, Khadilkar VV, Stanhope R, 1999 Pituitary dysfunction, morbidity and mortality with congenital midline malformation of the cerebrum. Eur J Pediatr 158: 97-102.
25. Lamesch C, Neumann S, Pfäffle R, Kiess W, Paschke R, 2002 Adrenocorticotrope deficiency with clinical evidence for late onset in combined pituitary hormone deficiency caused by a homozygous 301–302delAG mutation of the PROP1 gene. Pituitary 5: 163-168.
Address for correspondence:
Stefanie Petzold, University of Leipzig, Hospital for Children and Adolescents, Research Laboratory,
Oststrasse 21–25, 04317 Leipzig, Germany, Tel.: +49 (0)341 9726100, Fax: +49 (0)341 9726068,
e–mail: stefanie.petzold@medizin.uni–leipzig.de
Received 12-01-08, Revised 15-03-08, Accepted 10-04-08