1Department of Health’s Sciences, University of Florence, 2Clinical Ophthalmology Unit; Anna Meyer Children’s University Hospital; Florence, Italy
INTRODUCTION: Vernal keratoconjunctivitis (VKC) is a chronic conjunctivitis that mainly affects children living in temperate areas. The notable difference between genders and VKC’s resolution with puberty have persistently suggested a role of hormonal factors in VKC development. OBJECTIVE: To describe six cases of males with VKC and growth hormone deficiency (GHD) reported as a long-term follow-up during rhGH treatment.
METHODS: Six consecutive male patients (median age at GHD diagnosis 9.7, range 7.9 to 13.1 years) with VKC, were recruited from July 2005 to July 2013 at the Paediatric Endocrinology Unit of Anna Meyer Children’s Hospital in Florence, Italy. In these patients, anthropometric data were collected periodically. In three of these patients, data were collected to near-adult or adult height.
RESULTS: Familial history was uneventful for all patients. The target height was normal, ranging from 0.65 standard deviation scores (SDS) to 2.01 SDS. The patients showed a normal birth-weight (from -1.21 to 1.35 SDS) and birth-length (from -0.93 to 1.21 SDS). At GHD diagnosis, all of the patients exhibited demonstrated important growth retardation (from -2.05 to -2.78 SDS). Plasmatic concentrations of IGF-1 and IGFBP-3 were low (from -1.85 to -3 SDS and from -1.81 to -2.76 SDS, respectively). GH stimulation tests showed classic GHD symptoms in all of the patients. Pubertal onset was normal. All of the patients treated with rhGH responded well to rhGH treatment. Adult height, evaluated in three patients, was in accordance with their target height.
CONCLUSIONS: To our current knowledge, we have described for the first time six patients affected by VKC with GH deficiency, in some of whom we performed a long-term follow-up to adult height. Further studies will be needed to establish whether GHD may be a common feature of VKC patients. Nevertheless, it appears to be useful to carefully follow statural growth of VKC patients, while the possibility of a GH deficiency must to be taken into account in the presence of growth failure.
Children, Growth, Growth hormone, Growth hormone deficiency, Vernal keratoconjunctivitis
INTRODUCTION
Vernal keratoconjunctivitis (VKC) is a severe inflammatory disease that appears in children and adolescents with seasonal recurrence or, less frequently, as a perennial, persistent disease.1 The male-female ratio is from 4:1 to 2:1; the prevalence of the disease is 7.8/100,000 and the incidence is 1/100,000.2,3 VKC pathogenesis involves various factors, including environmental allergens, climate, and genetic predisposition.1,2 Although VKC has always been considered an allergic disorder, its aetiology and immunopathogenesis remain unclear.1 A large proportion of patients have no familial or personal history of atopic disease and can have negative results on the standard allergy diagnostic tests.1 Through inflammatory endocrine-immune stimuli, eosinophils, conjunctival fibroblasts, epithelial cells, mast cells, and TH2 lymphocytes provoke multiple dysfunctions in conjunctival biochemistry and histology.4 Cytological, biohumoral, immunohistological, and molecular biological studies indicate that VKC is a TH2 lymphocyte-mediated disease.2
Although VKC usually lasts from 4 to 10 years and resolves after puberty, it can still be present and even worsened in some adult patients (about 0.5%). The notable difference between genders and VKC’s resolution with puberty are features that have persistently suggested a role of hormonal factors in the development of VKC.2
In fact, hormones and neuroendocrine factors may influence the immune deviation and the response to environmental factors in addition to unknown genetic predisposing conditions.2 In particular, although there is a strict crosstalk between the immune and nervous system, whether neurotransmitters and neurotrophins may influence conjunctival inflammation is currently unclear.
The purpose of this study was to describe six cases of male patients with VKC and GHD reported as a long-term follow-up.
SUBJECTS AND METHODS
Data were collected from six patients (6 males, median age at the diagnosis of GHD 9.7, range 7.9 to 13.1 years) with VKC, recruited consecutively from July 2005 to July 2013 at the Paediatric Endocrinology Unit of Anna Meyer Children’s Hospital in Florence, Italy.
Ethical approval was obtained from the Ethics Committee of Anna Meyer Children’s Hospital. After explaining the nature of the study, written informed consent was obtained from the parents or the patients according to the age and patients’ ability to give assent.
Case definition and study protocol
VKC diagnosis was established according to the patient’s history, clinical signs and symptoms, skin test results, and determination of serum total and specific IgE. VKC diagnosis was based on the presence of recurrent symptoms of itching, photophobia, and tearing in early spring. The symptoms were associated with a mild to severe cobblestone-like appearance in the upper tarsal conjunctiva, mucous discharge, epithelial keratopathy, and at least two eosinophils per optic field (x100/oil immersion) in the conjunctival scraping.
Ocular signs were evaluated as recently reported. In all of the patients, a brief, subjective clinical activity score (0-10) was assigned considering the overall disease severity (0 = no symptoms and no active signs; 10 = very severe symptoms and signs). Additionally, an individual score for corneal involvement was assigned: 0 = clear cornea; 1 = local punctate keratitis; 2 = diffuse punctate keratitis; 3 = focal confluent de-epithelisation; 4 = corneal shield ulcer. All patients underwent skin-prick testing and serum determination of specific IgE for common inhalant and food allergens (UniCAP Specific IgE; Pharmacia, Uppsala, Sweden); patients were considered to be IgE sensitised with at least 1 positive skin-prick test response or when serum-specific IgE determination (>0.1 kU/L) was present.
A possible prolonged administration of systemic or topical drugs such as antihistamines, corticosteroids, cyclosporine, and nonsteroidal anti-inflammatory drugs was also investigated.
In all subjects, biochemical and endocrine examinations were performed to rule out possible causes of growth failure. At the time of the auxological evaluation, all of the patients showed a height less than 2 standard deviations (SD) below the mean for the normal population and/or a subnormal growth velocity. In all of the subjects, we studied the GH response to oral clonidine, arginine, and/or GH releasing hormone (GHRH) + arginine tests.
GHD diagnosis was carried out based on auxological criteria (height and/or growth velocity 2 SD below the mean for the normal population) and a GH peak below 10 μg/L (below 20 μg/L after GHRH + arginine test) after two provocative tests on two separate occasions.5 Anthropometric data were collected periodically (approximately every 6 months to 1 year). Near- and/or Final Height (FH) was commonly defined, recognising that children may continue to grow after achieving this definition, for patients in whom both chronological age (CA) and bone age (BA) were at least 14 yr. for females and 16 yr. for males and with a HV <1 cm/year for two consecutive years.6 Intracranial imaging was obtained by magnetic resonance imaging (MRI) using precontrast coronal spin echo T1-weighted images followed by postgadolinium T1-weighted imaging.
Height, height velocity (HV), and weight were measured, and body mass index (BMI; weight in kilograms/height in square meters) was calculated in all patients. Height, HV, and BMI values were converted to standard deviation scores (SDS). SDS was calculated according to the following formula: (patient value mean of age-related reference value)/standard deviation of the age-related reference value.6 Height, height velocity, and weight were evaluated according to Italian growth references.7 Pubertal stage was assessed according to Marshall and Tanner8 and testicular volume was determined with the Prader orchidometer.
BA was evaluated through radiographs of the left hand and wrist and then calculated according to the Greulich and Pyle method.9 TH was estimated according to the method of Hermanussen and Cole.10 Mid-parent height was calculated as an SDS and correcting this by a factor corresponding to the influence of assortative mating and parent-offspring correlations.6
The GH stimulation tests (clonidine, arginine, insulin) were started between 08.00 and 08.30 after an overnight fast (only water was allowed). A plastic cannula was placed in a large vein and kept open by saline solution for serial blood sampling.
RESULTS
The main auxological features of the patients are summarised in Table 1.
One patient (16.6%), a male (patient 2), has a positive family history for short stature (mother’s height -2.1 SDS). Familial history was uneventful for all of the subjects. Three patients (50%) came from only-child households of non-consanguineous, healthy, Italian parents. One male, the second child, has an older sister with final height (-0.54 SDS). The other two males have younger brothers with normal growth. All of the parents have no positive history for pubertal delay except for the mother of patient 1; she had a menarche at the age of 14.4. The target height was normal in all of the families, ranging from 0.65 SDS and 2.01 SDS.
The evaluation of drug use that could possibly have influenced statural grow-h showed that one patient, a male, has a positive history for a prolonged corticosteroids use in childhood.
All of the patients were born by at-term uncomplicated gestations. The patients had a normal birth-weight (from -1.21 to 1.35 SDS), birth-length (from -0.93 to 1.21 SDS), and head circumference (from -0.42 to 0.65 SDS).
At VKC diagnosis, all of the patients demonstrated normal-subnormal growth (from -1.23 to -1.84 SDS); even if significantly lower than the respective target height. GHD diagnosis was delayed (from 8.99 to 11.95 years); at the first paediatric endocrinological evaluation, auxological data showed that all patients presented important growth retardation (from -2.05 to -2.78 SDS).
At the time of GHD diagnosis, the bone age, evaluated by Greulich and Pyle’s method, revealed almost all patients as being considerably delayed (from -2.1 years to -3.4 years respect to the chronological age). Only patient 5 exhibited a non-significantly delayed bone age.
Growth hormone stimulation test evaluation revealed a classic GHD in all of the patients. Plasmatic concentrations of IGF-I and IGFBP-3 for age and sex were low in all of the patients (from -1.85 to -3 SDS, and from -1.81 to -2.76 SDS, respectively).
All patients, with the exception of patient 6 (a new diagnosis who had not started the rhGH treatment; Figure 1.6) were treated with rhGH. None of the patients showed multiple pituitary hormone insufficiencies.

Figure 1. Part A. Growth charts of the six patients. Dashed and continuous lines represent the centile lines. The black dots depict a single height determination of each patient. The black arrows indicate the age of beginning of growth hormone treatment. TH represents the target height (mean ± 6.5 cm).
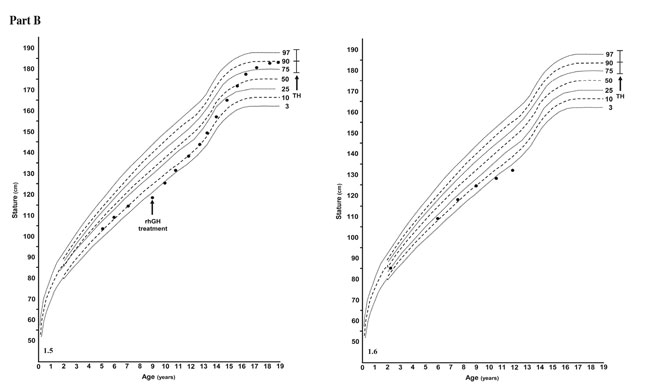
Figure 1. Part B. Growth charts of the six patients. Dashed and continuous lines represent the centile lines. The black dots depict a single height determination of each patient. The black arrows indicate the age of beginning of growth hormone treatment. TH represents the target height (mean ± 6.5 cm).
The auxological follow-up disclosed that all of the rhGH treated patients had a very good response to the GH therapy (Figure 1.1 to 1.5).
Pubertal onset, pattern and development were in the normal range. The final height, evaluated in three patients, was in accordance with their target height.
DISCUSSION
In this report, to the best of our knowledge, we have described for the first time six patients affected by VKC accompanied by GHD, and we performed a long-term follow-up to adult height in three of these patients. In these patients, the GH therapy was effective (Figure 1.2, 1.3, and 1.5), enabling the achievement of a final height corresponding to their target height.
There are no data evaluating the growth pattern in patients with VKC. Thus, further studies will be needed to determine whether a GHD may be a common, frequent feature of these patients. In addition, other data are necessary to study whether VKC patients without GHD may reach an adult height in accordance with or below their target height. Nevertheless, our study suggests carefully following the statural growth of VKC patients, and, in those presenting growth retardation and/or short stature, determination of the presence of a GHD.
Even if the relationship between VKC and GHD remains unknown, VKC pathogenesis seems to indicate a role of hormonal factors in their development.2 As stated above, several authors have documented endocrine disorders in 67% of VKC patients. These disorders include not only gonadal insufficiency, but also disorders of the hypophisis or of the hypothalamic-hypophisiary complex.11 In fact, a role for sex hormones has been postulated in the pathogenesis of this disease. This assumption derives from the prevalence of VKC in males versus females and the spontaneous disease resolution at puberty. Certainly, sex hormones may play a relevant role in the pathophysiology of allergic diseases. Oestrogen and progesterone have been shown to be active players in the ocular immune system, and previous immunohistochemical studies have demonstrated that oestrogen and progesterone receptors were overexpressed on the conjunctiva in VKC, exerting a proinflammatory effect through the recruitment of eosinophils to the conjunctival tissue.12
However, sex hormone related diseases such as gynaecomastia, polycystic ovary syndrome, mammary fibroadenoma, adiposogenital dystrophy, and autoimmune diseases as well as tendency to develop autoimmune disorders were reported by 2% and 8% of patients suffering with VKC.13,14 For example, we have revealed (unpublished data) a higher frequency of thyroid disorders, such as subclinical hypothyroidism and/or thyroid autoimmunity, in VKC patients, though thyroid function was normal in all of our patients reported in this study.
The inflammatory pattern of VKC may also explain the tendency to develop GHD in some of these patients. For example, some data point to the presence of high values of TGF-beta15 or IL-17.16 IL-17 also plays an important role in many autoimmune diseases,16 while Growth Hormone Releasing Hormone (GHRH) has been demonstrated to play an immunoregulatory role in the secretion of IL-17 in human peripheral blood mononuclear cells.17
The reasons for the presence of GHD in some patients with VKC have not been clarified and this association may be fortuitous. However, our study strongly suggests the need for future studies of growth hormone secretion in patients with VKC.
Nevertheless, it is interesting to note that an excessive growth of eyelashes (trichomegaly) has been reported both in patients with VKC18 and in those with some congenital conditions. Familial trichomegaly has been described19 and rare cases of congenital trichomegaly with pigmentary degeneration of the retina and growth retardation were also reported.18
In conclusion, we suggest that the short stature in VKC could be due to the presence of GHD, though further studies are necessary to look into endocrine problems and to better understand growth in VKC patients. Since there are currently no data that evaluate the growth pattern in patients with VKC, additional investigation is clearly necessary to determine whether a GHD may be a common or frequent feature of these patients. Moreover, other data are required to examine whether VKC patients may reach a final height in the normal range or below their target height. Meanwhile, it also appears to be important to carefully follow the statural growth of these patients, and the possibility of a GHD must additionally be considered.
FINANCIAL COMPETING INTEREST
The authors have no financial and non-financial competing interests in relation to this manuscript.
REFERENCES
1. Leonardi A, 2002 Vernal keratoconjunctivitis: pathogenesis and treatment. Prog Retin Eye Res 21: 319-339.
2. Leonardi A, Secchi AG, 2003 Vernal keratoconjunctivitis. Int Ophthalmol Clin 43: 41-58.
3. Leonardi A, Busca F, Motterle L, et al, 2006 Case series of 406 vernal keratoconjunctivitis patients: a demographic and epidemiological study. Acta Ophthalmol Scand 84: 406-410.
4. Ono SJ, Abelson MB, 2005 Allergic conjunctivitis: update on pathophysiology and prospects for future treatment. J Allergy Clin Immunol 115: 118-122.
5. Bercu BB, Shulman D, Root AW, Spiliotis BE, 1986 Growth hormone (GH) provocative testing frequently does not reflect endogenous GH secretion. J Clin Endocrinol Metab 63: 709-716.
6. Stagi S, Galli L, Cecchi C, et al, 2010 Final height in patients perinatally infected with the human immunodeficiency virus. Horm Res Paediatr 74: 165-171.
7. Cacciari E, Milani S, Balsamo A, et al, 2006 Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J Endocrinol Invest 29: 581-593.
8. Tanner JM, Whitehouse RH, 1976 Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch Dis Child 51: 170-179.
9. Greulich WW, Pyle S 1959 Radiographic Atlas of Skeletal Development of the Hand and Wrist. In Stanford, CA: Stanford University Press.
10. Hermanussen M, Cole J, 2003 The calculation of target height reconsidered. Horm Res 59: 180-183.
11. Fiero del Río 1948 Estudios endocrinológicos en la conjuntivitis primaveral. Anales de la Sociedad Mexicana de Oftalmología XXII: 54-71.
12. Bonini S, Lambiase A, Sgrulletta R, Bonini S, 2003 Allergic chronic inflammation of the ocular surface in vernal keratoconjunctivitis. Curr Opin Allergy Clin Immunol 3: 381-387.
13. Bonini S, Bonini S, Lambiase A, et al, 2000 Vernal keratoconjunctivitis revisited: a case series of 195 patients with long-term followup. Ophthalmology 107: 1157-1163.
14. Tesse R, Spadavecchia L, Fanelli P, et al, 2010 Treatment of severe vernal keratoconjunctivitis with 1% topical cyclosporine in an Italian cohort of 197 children. Pediatr Allergy Immunol 21: 330-335.
15. Leonardi A, Di Stefano A, Motterle L, Zavan B, Abatangelo G, Brun P, 2011 Transforming growth factor-β/Smad - signalling pathway and conjunctival remodelling in vernal keratoconjunctivitis. Clin Exp Allergy 41: 52-60.
16. Zicari AM, Nebbioso M, Zicari A, et al, 2013 Serum levels of IL-17 in patients with vernal keratoconjunctivitis: a preliminary report. Eur Rev Med Pharmacol Sci 17: 1242-1244.
17. Stepien T, Lawnicka H, Komorowski J, Stepien H, Siejka A, 2010 Growth hormone-releasing hormone stimulates the secretion of interleukin 17 from human peripheral blood mononuclear cells in vitro. Neuro Endocrinol Lett 31: 852-856.
18. Pucci N, Novembre E, Lombardi E, et al, 2005 Long eyelashes in a case series of 93 children with vernal keratoconjunctivitis. Pediatrics 115: e86-91.
19. Harrison DA, Mullaney PB, 1997 Familial trichomegaly. Arch Ophthalmol 115: 1602-1603.
Address for correspondence:
Stefano Stagi, E-mail: stefano.stagi@yahoo.it
Received 08-10-2013, Accepted 03-12-2013