Department of Nutrition and Food Science, Faculty of Biology, University of Barcelona, Barcelona, Spain, and CIBER Obesity and Nutrition, Institute of Health Carlos III, Spain
Androgens, Estrogens, Glucocorticoids, Metabolic syndrome, Obesity, Ponderostat
SEXUAL DIMORPHISM IN THE METABOLIC SYNDROME
The clairvoyant early implication of sex hormones in the characterization of the metabolic syndrome (MS) was detected early,1 and in accordance with the well-known sex-related main patterns of fat deposition in obesity: gynoid and android.2 The differences point to a direct implication of androgens and estrogens in the development, properties and maintenance of obesity and, by extension, to the cumulus of diseases grouped in the MS.3,4 For a long time, the key issue of the MS, i.e. the metabolic event explaining (and justifying) most of the derangements of the MS, has been considered to be insulin resistance.5,6 Later, the emphasis was directed to a lower molecular level, the inflammatory response of tissues, mainly white adipose tissue (WAT);7 this response was assumed to be mediated principally by cytokines.8 Currently, however, the number of research fronts open is considerable, with the emphasis on mechanistic molecular-level analyses.9 The widespread focus on the signalling pathways to account for the modulation of physiological changes and the commencement of pathological derivation10,11 has, to a certain degree, caused us to lose some fundamental perspective. The processes at the molecular level never occur in an ‘ideal’ isolated setting, but are intricately involved in multiple metabolically and structurally complex systems, i.e. in cells which communicate with other cells within the same tissue, organ or at a systemic inter-organ level. There are several layers of superimposed (and completely intergraded) regulatory systems which contain fail-safe mechanisms to prevent catastrophic events in order to efficiently maintain life.
When we analyse the MS from a strictly physiopathological point of view, the most striking effects observed are those defined in the deadly quartet: obesity, hypertension, insulin resistance and hyperlipidemia.12 However, in this context, the term obesity is often further defined as upper body or visceral obesity.13 This is an important point. Many obese women show a fairly normal metabolism, lacking a full manifestation of the MS,14,15 but in a high proportion of men, visceral obesity, even of limited extent, is often accompanied by the full manifestation of the MS associated diseases.16 There is obviously a significant number of obese individuals who simply store more fat in their bodies without significant inflammation, insulin resistance, hypertension etc.;15,17 this type of obesity may reach high proportions of body fat, but it is often deposed in a lower body fat deposition (largely subcutaneous) pattern.17 In contrast, android obesity is more harmful, even with markedly lower proportions of body fat.18 It is obvious that fat storage alone cannot constitute the basis of the MS pathology.19 Indeed, the reverse is true, since there are individuals with all the symptoms of a developed MS who show a body fat content within normalcy and who are oxymoronically defined as normal-weight obese.20,21
In general, women tend to show a dispersed distribution of fat most of which is located in the subcutaneous space, whereas in men a large part of the fat is concentrated within the abdominal cavity.22 In rodents, the differences are less marked, since a considerable amount of fat is stored in the abdominal space in the retroperitoneal and perigonadal pads, which cannot be equated with the omental fat of humans;23 it is hypothesized that rat mesenteric fat is closer in pathogenic potential and metabolic activity to human visceral fat.24
In any case, we should distinguish two main types of obesity, android and gynoid, based on the distribution of fat, the overall metabolic transcendence of excess fat and the severity of its health consequences. The implication of steroid hormones characterizes this large difference.25,26 There is a high degree of superposition between the two main types of fat distribution, but in most cases the consequences can easily be derived from changes in sex hormone availability.27
Estrogens tend to limit body fat deposition,28,29 and when there is ample fat storage under their aegis, its distribution adopts the gynoid pattern.30,31 At menopause, when estrogen synthesis and levels sharply decrease, a redistribution of body fat occurs, diffusing the contours of the gynoid pattern towards a hybrid android-influenced pattern.32 This change has been attributed to a shift in the imbalance between androgens and estrogens.33
Androgens facilitate muscle and bone growth and maintenance34,35 and are also assumed to promote the male-related android distribution of body fat.36 However, testosterone diminishes the accumulation of intra-abdominal fat.37 In fact, visceral fat is a major site for androgen inactivation,38,39 thus the fight is two-sided. In the MS, testosterone administration tends to lower body fat,40 and hypoandrogenism41 or androgen deprivation treatments are correlated with marked increases in fat deposition, both visceral and subcutaneous.42
Hypoandrogenism (i.e. low levels of serum testosterone) is a common finding in the MS,41,43 usually paired with decreased dehydroepiandrosterone (DHEA) levels44 and lower synthesis/levels of androstenedione and testosterone.45,46 Serum testosterone activity may be even lower because of its binding to SHBG 47,48 (sex hormone-binding globulin) and other testosterone-binding proteins,49 including CBG50 (glucocorticoid-binding globulin). The generalized hypoandrogenemia observed in male obesity51 is compounded by an increased aromatization of androgens to estrogens, especially in the enlarged adipose tissue52 of the obese individual, producing estrogens53 and thus inducing estrogenization. Obesity favours this decrease in androgens by activation of the expression of aromatase,54 which adds to the large mass of adipose tissue, but also because of the targeted destruction of androgens.38,39,50 Inhibition of aromatase in obese men helps reverse their altered hypothalamus-pituitary-gonadal axis towards androgen normalization.55
STEROIDS AND MS
Role of androgens in the pathophysiology of MS
Our blueprint for development sets the timing for the establishment of a number of changes in body function, shape and patterns of activity and behaviour inherited from our ancestors. The sex-related differences in fat distribution have a strong behavioural and sexual component; they have been well described for groups of gorillas,56 but are also—in part—applicable to humans. These changes (body shaping, baldness and/or graying hair in males) occur slowly, following a gene-controlled pattern 57 over a prolonged period of time, at specific points of the life-cycle and parallel to sex hormone synthesis (and levels).58,59 The process is controlled by the brain, mainly through hypothalamic control of gonadotropin secretion by the hypophysis, which regulates androgen and estrogen levels and follows precisely established developmental patterns based on genetics60 and seasonal and circadian rhythmicities.61 The timing and extent of changes are also regulated by energy availability (e.g. by leptin secretion by WAT stores62,63) and other environmental factors (e.g. through cortical signals, glucocorticoids, etc). Nevertheless, they always result in long or very long-term changes following the evolutionary blueprint for reproductive effectiveness and group survival.
The implication of estrogens and androgens in the development of the diseases associated with the MS was recognized very early, even before the term inflammation was applied in this context.31 A telltale sign of their implication is the growing incidence of the polycystic ovary syndrome,64 largely due to an alteration in androstenedione metabolism,65 but characterized by insulin resistance,66 low adiponectin67 and a number of MS-related pathological traits.68-70 Polycystic ovary syndrome is a component of the MS.71,72 Central to polycystic ovarian syndrome is an altered metabolism of androgens.73-75 Polycystic ovary syndrome is usually treated with metformin,76 an insulin sensitizer not affecting androgen metabolism.
It has been established that the different pathological traits of the MS are present with varying intensity in different individuals, with predominance of diabetes, atherosclerosis, obesity, etc., often with non-pathological degrees of some of the diseases of the syndrome, although in some cases the full spectrum of diseases is evident. Polycystic ovary syndrome may be one such MS manifestation,72 this hypothesis largely based on its superimposed symptoms.77-79 It should be noted here that MS affects not only the female reproductive system but also the male. Indeed, MS in males causes hypoandrogenism,41,80 with decreased testosterone81,82 as well as adrenal weak androgen dehydroepiandrosterone DHEA levels.83,84 Thus, in both sexes the MS is associated with altered androgen metabolism.80,85-87
The imbalance between lower androgens and raised corticosteroids signals a permanent shift in metabolic priorities: androgens are highly anabolic, increasing protein preservation and skeletal (muscle, bone) growth,88-90 whilst glucocorticoids promote protein degradation (to fuel gluconeogenesis, thus maintaining euglycemia) and induce skeletal regression, including loss of minerals from bone and generalized wasting.91-93 Androgens are natural antidepressants,94-96 and excess of glucocorticoids is associated with depression,97-99 another example of the antagonistic roles these steroid hormones play, at least in the brain, which in fact controls everything else in all physiological mechanisms.
Role of estrogens in the pathophysiology of MS
To produce estrogens we need first to synthesize androgens, starting with dehydroepiandrosterone; the last step being aromatization100,101 (Figure 1). Thus, the decline of DHEA caused MS may in the end diminish the estrogen-mediated beneficial effect on vascular endothelium integrity102 and defence against superoxidation.103 Estrogens also ameliorate insulin resistance104 and facilitate the handling of low-density lipoproteins by the liver.105,106 Estrogens play a protective role in the brain,107,108 the vascular tree109,110 and indeed in most other tissues, which is compatible with their profound effects on fat metabolism.111,112 The decline of estrogen levels following menopause113 results in a rapid weakening of these protective roles, altering the equilibrium with androgens, especially in individuals not on estrogen-replacement therapies.114 This is not akin to androgenization, since the decline of estrogens is followed by a fall in androgens. Estrogen replacement therapy may indeed deepen the deficit of androgens115 by stimulating shared inactivation paths. However, the loss of sex steroids equilibrium seems to be mainly an imbalance of both androgens and estrogens against corticosteroids. The decline in estrogen levels may well be the result of the loss of androgens.113

Figure 1. Metabolism of steroid hormones. Relationships between the main glucocorticoids, androgens and estrogens.
The ponderostat hypothesis
Estrone is a powerful anabolic hormone, promoting growth and the deposition of fat in the young,116 but is a fairly inactive estrogen.117 Estrone is synthesized in significant amounts by WAT,100,118 which stores it largely as its oleoyl-ester,119,120 a precursor of a postulated ponderostat signal.121
The ponderostat system (Figure 2) regulates lipid storage to an optimally adjusted mass, but the MS rapidly alters the pre-established ponderostat settings in such a way that either the brain becomes less sensitive to the signal(s) from WAT or this tissue shows a decreased capability to synthesize the signalling molecule(s). In any case, we may say that in obesity, the brain has lost its ability to control the size of WAT,122 probably because of inadequate sensing of adipose tissue actual size due to either lower levels or insensitivity to ponderostat signals. The case of the active form of oleoyl-estrone is paradigmatic, since most of the damages caused by the MS are decreased or minimized by the pharmacological administration of this oleoyl-estrone, derivative and postulated precursor of a ponderostat signal.123
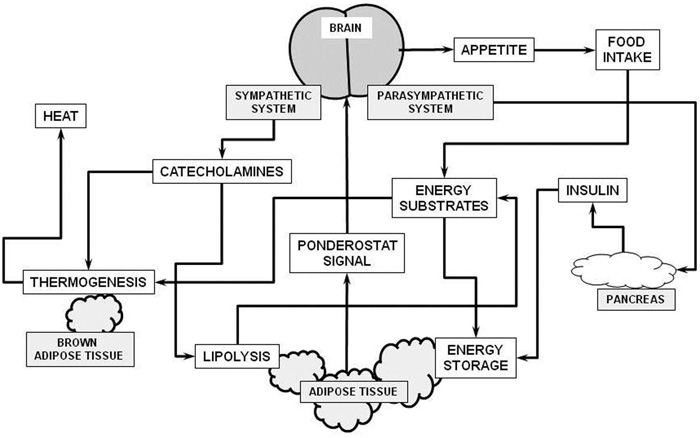
Figure 2. Schematic representation of the
ponderostat model. The “ponderostat signal” is released into the bloodstream
proportionally to the mass of energy stored in adipose tissue. The brain
recognizes its level and associates it to age, sex and energy availability. If
the amount of fat reserves is inadequate, the brain can increase the
availability of substrates largely through control of appetite (and thus food
intake) and the action of the autonomous nervous system. A perceived lack of
energy increases food intake and the secretion of insulin, resulting in an
overall increase in energy storage in adipose tissue. Conversely, perceived
excess energy stored in adipose tissue elicits a decrease in food intake, but
also a sympathetic response, increasing lipolysis. In this manner, a larger
proportion of available energy substrates is endogenous and may be used by brown
adipose tissue (and other organs/tissues) for thermogenesis and its final
elimination as heat.
Ponderostat settings are subjected to the changes in body weight reserves pre-established by our genes, epigenetic modulations and developmental influences, and shift throughout our entire life. One of the most dangerous effects of the MS is precisely the changes it induces in our ponderostat settings. Obese individuals encounter all types of physiological difficulties when trying to lose weight124-126 largely because their ill-adjusted ponderostat setting is defended by the body mechanisms that prevent the loss of its reserves. Our lack of knowledge on how the ponderostat sequence of events works and how it is set up makes us uncertain as to the importance of this mechanism with regard to MS manifestations.127 Glucocorticoids may play a role in the maladjustment of the ponderostat setting, both because of their omnipresence in the MS128-130 and their role, as yet little known, in the control of androgen and estrogen metabolism,131-134 which may extend to its counteracting effects upon the estrone-derived ponderostat signal candidate.135
Role of glucocorticoids in the pathophysiology of MS
It is clear that androgens and estrogens cannot alone account for the ravages and, especially, the timing of appearance of the associated pathologies of the MS. However, there is a third and critical element in the equation: corticosteroids, and mainly glucocorticoids. The implication of glucocorticoids in obesity are well known.136-138 Specifically, the main enzyme converting cortisone (less active) and cortisol (active), type 1 11β-hydroxysteroid dehydrogenase, is present and functional in WAT139,140 (Figure 1). This allows adipose tissue to generate active cortisol from circulating less active cortisone.141,142 Cortisol is essentially the product of the cortex of the adrenal glands, which also produce aldosterone,143 dehydroepiandrosterone (DHEA)144 and lesser amounts of sex hormones.145 Glucocorticoids play a crucial role in the control of homoeostatic stability: they induce resistance to insulin146,147 and other hormones and cytokines135,148 (including glucocorticoids themselves149) and enhancement of liver gluconeogenesis,150 largely at the expense of body protein;151 they also mobilize minerals from the bone,152 hamper the full immune response to real (or feigned) aggressions,153 favour lipid deposition147 and help the body to recover after any physical exertion or stressful situation (exercise, exhaustion, infection, stress, etc.).154,155 These roles are critical for survival and thus take precedence over the more slow effects of sex hormones, which effects become more extended over time.156 The immediacy of glucocorticoid action, however, is not absolute, especially when we compare it with actions as fast as these of catecholamines whose effects start in the split-second range (i.e. fight-or-flight situations);157 glucocorticoids act at a slower pace, but nevertheless much faster than the longer periods needed for many of the actions of estrogen and androgen to manifest.158,159
In the MS, the hypothalamus-pituitary-adrenals axis is more active than under normal conditions160 and there is a close relationship between obesity and stress.161,162 However, the circulating levels of cortisol in MS are often within the normal range,163 since part of the modulation of corticosteroid action depends on the peripheral conversion (i.e. within the same target tissue) of cortisone and of the less active cortisone.164,165 There is also a complex control of hormone availability through variability of both its transport in blood compartments and attachment to cells largely through CBG.166,167
An additional third level of regulation arises from the formation of dimeric structures by steroid hormone receptors,168,169 which enables the chimeric formation of hybrids of glucocorticoid-bound receptors with other ligands such as androgens,170 estrogens171 and retinoids.172
Glucocorticoids inhibit the synthesis and activity of androgens.173-175 However, androgen precursor DHEA (and, consequently, precursor of estrogens176) has anti-glucocorticoid activity.177,178 Glucocorticoids may hamper the activity of estrogens. 179 It should be noted here that activation of the estrogen receptor affects the brain glucocorticoid receptor-dependent effects in the amygdala and in the neuroendocrine system are opposed.180 However, glucocorticoids and estrogens may also act synergistically. Indeed, estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression and suppress vascular endothelial inflammation.181,182
The glucocorticoid relationship with androgens is perhaps more antagonistic because of their different effects at the cellular level: androgens as enhancers of protein synthesis are powerful anabolic agents,183 and corticosteroids tend to mobilize protein184,185 to reap enough gluconeogenic substrates for the maintenance of glycemia.186 Excess glucocorticoids (often at pharmacologic levels) induce protein, glycogen and mineral wasting.92
In the climacteric transition, women’s synthesis of estrogen decreases187 and a few androgenic effects appear, such as the redistribution of body fat188,189 and weight increase190 as well as the loss of most of the protective effects of estrogen on the brain, skin and cardio-circulatory systems.191,192 There is a relative increase in androgens at menopause,193,194 but glucocorticoid activity is also increased.195 The transformation in body fat redistribution and increased deposition during menopause appears to be a consequence of a transient androgen increase and a slow but steady rise of the circulating glucocorticoids.
One of the main health problems associated with menopause, bone demineralization,197 is a telltale indicator of both low estrogens and high corticosteroids, especially the latter. In males, this demineralization occurs at a slower pace, even in obese individuals with low levels of circulating testosterone.198 It seems that even low androgen levels may be sufficient to prevent or limit osteoporosis. Consequently, it is not logical to attribute the change of gynoid to android (or intermediates) fat distribution patterns at menopause to androgenization. If there were indeed sustained androgen activity increases, the bone may be effectively protected as in men. Meanwhile, the conversion of androgens to estrogens, albeit limited, should help further protect bone minerals. The effects observed in post-menopausal women are not, thus, only attributable to an imbalance between estrogens and androgens but mainly between estrogens and glucocorticoids.
GLUCOCORTICOIDS, INFLAMMATION AND MS
The deranged actuation of the immune system may also be at the root of a number of MS-related diseases such as psoriasis199,200 and other autoimmune pathologies.201 Probably, the excess proinflammatory cytokine production of inflamed cells induces an excessive stimulation of the defence system.202,203 The actual absence of real enemies of the immune system to efficiently fight, as well as the lack of coordination of the estrogen and glucocorticoid secretion and function, may enhance the levels and activity of the latter. The higher incidence of a few types of cancer (excluding those related to food consumption204) in obesity and the MS205,206 corroborate this interpretation.
The change in microbiota composition elicited by excess secretion of nitrite into the digestive system207 may also combine with a strong and activated immune system208 to modify the intestinal interface with the microbial symbiotic biota.209 This question has barely been analysed, except for the verified differences in the species distribution of the microorganisms, confirming what is known about obese microbiota,210,211 which is fairly independent of the type of food consumed212 and is very different from the normal microbiota in species distribution and proportions. The existence of low, albeit constant, levels of lipopolysaccharide in the blood of obese individuals213 suggests that a low-level intensity microbial-induced inflammation may be a significant cause of the inflammatory response observed in the MS.214 However, there are other plausible explanations, such as the great predominance of glucocorticoids, secreted in response of an enhanced immune system activity,153 acting on the control of the bacteria kept at bay in the intestine,215 or an excessive monocyte and macrophage phagocytic response to infection releasing residual lipopolysaccharide into the bloodstream.213
BRAIN PONDEROSTAT SETTINGS AND MS
The most elusive (and least known) effects of the MS occur in the brain; they can be arbitrarily divided into a number of superimposed actions:
a)modulation of the ponderostat size of body fat
reserves (changes in ponderostat setting, modulation of food intake and
modulation of energy expenditure)
b)effects on rhythms, sleep and meal
patterns
c)effects on thymic and reward circuits
d)modulation of hormone secretion
e)changes in the pattern (blueprint) for the
life-cycle
f)alterations of behaviour
Evidently, the modification of the ponderostat setting is a powerful tool for the epigenetic adaptation to environments with low food availability, since a higher setting results in higher efficiency in food energy utilization and the storage of fat when the energy is available. This trait (thrifty phenotype216,217) is a direct cause of obesity under conditions of excess available food energy. However, the consumption of a very rich diet for a long time (e.g. cafeteria diet218,219) also disrupts the adjustment of the ponderostat and results in obesity220 without the epigenetic preparation for hard times ahead. It may be speculated that the ponderostat setting may be altered by metabolic or hormonal signals. It is difficult, however, to determine how, since we do not know for certain which is (are) the adipose tissue signal(s) that inform the brain of the mass of stored energy.
Obese persons retain a considerable ability to maintain their body weight,221,222 albeit in a high, non-physiological setting. Hypocaloric diets have a limited effect in the obese223,224 because in the end the body tries to regain the preset mass of reserves225 and the ponderostat system forces the metabolic paths to save energy and store it to maintain the pre-established levels (ponderostat setting). The ponderostat works mainly by establishing an equilibrium between energy intake (appetite) and energy expenditure (complemented by the modulation of thermogenesis)226 (Figure 2). Appetite is largely controlled by the hypothalamus through two confronted systems: NPY as main food intake promoter227,228 and melanocortins as inhibitors.229,230 Significantly, the precursor of melanocortins, proopiomelanortin, is also a precursor of endorphins and ACTH corticotrophin.231 Melanocortin signaling is also related to melanin deposition in the skin232 (it intervenes in the development of acanthosis nigricans,233 a complication of diabetes234). Melanocortin is closely functionally related to orexins235 which intervene in the establishment of the complex network controlling food intake.236 Orexins are also implicated in the regulation of sleep and sleep cycles.237
OTHER ASPECTS OF THE BRAIN – ADIPOSE TISSUE INTERACTION
There is a direct relationship between sleep and the development of the MS.238,239 Sleep deprivation induces MS-like disturbances;240,241 in general, obese individuals sleep less time than non-obese242,243 and the deleterious effects of sleep apnoea are not only the consequence of the catecholamine surges but mainly of the loss of sleep quality.244 The sleep changes observed in the MS are also related to the alteration in circadian feeding and activity rhythms found in obesity245 and which are largely regulated by ACTH corticotropin / corticosteroids.246 The loss of rhythms favours increased (unscheduled) food intake and the nibbling of food results in a disarranged meal feeding structure,247 which adds to the behavioural and neurochemical alterations associated with the MS.
It is important to note that in a small area of the lower brain (hypothalamus and surrounding structures) a large part of the control of food intake is concentrated,248 including, most probably, the adjustment of the ponderostat setting, the control of thermogenesis, sleep and circadian rhythms and the regulation of the hypophysis, which in turn controls most of the endocrine axes that regulate the function of the thyroid, adrenals, gonads and the water, mineral and electrolyte equilibrium.249-251 These mechanisms should be considered together with the direct influence on the autonomous nervous system functions (control of digestive tract motility, thermoregulation, heart rate, arterial tension and hemodynamic changes, to cite a few).252-254
The centres controlling the nervous system mechanisms of reward (dopamine, endorphins)255 are close to and in direct relation with the hypothalamus,256,257 which is also under the more or less direct control of cortical (voluntary) and sensorial areas,258,258 including interoceptors detecting the status of intestine replenishment (and the nature of its contents),259 glycemia,260 blood acid-basic equilibrium, oxygen and osmotic pressure.261,262
Pharmacology gives us a number of possible clues to the function of the system. If cortical hedonic signals decrease by applying a pre-established hypocaloric diet, the increase of serotonin signalling helps reduce body weight (dexfenfluramine, sibutramine)263,264 in a process akin to the treatment of depression (fluoxetine, sertraline).265,266 Again, glucocorticoids counter these effects and induce depression,267 which may be counteracted by serotonergic stimulation.268 The implication of cannabinoid receptors269 (rimonabant is an inverse cannabinoid CB1receptor blocker270), and catecholamines (amphetamine inhibiting food intake271 and phentermine or ephedrine increasing energy expenditure272) shows that the mechanisms controlling food intake and energy output, i.e. the ponderostat-driven mechanisms to regulate body energy, are multiple, complex and intertwined.
There are a number of drugs that cause iatrogenic obesity, including synthetic glucocorticoids,273 adrenergic blockers274 and a number of drugs used for psychiatric disorders.275,276 The discontinuation of nicotine stimulation in smokers produces a significant increase in appetite and permanent weight gain277 and the improvement of a number of psychiatric conditions results, again, in overweight.278 Sometimes it It is difficult to distinguish between iatrogenic effects and normalization, i.e. suppression of a continued non-physiologic stimulus such as nicotine.
Surprisingly, the relationship of the MS with eating disorders is less manifest. A number of obese individuals present the psychological characteristics of binge eating,279,280 but this may be a result of a cooperative cause, overeating, that has brought them to obesity,281 scaled up to the MS as a consequence, not the other way around.282 This may also be a consequence of unchecked stress that is compensated by comfort feeding.283,284
In sum, there is considerable evidence linking the different diseases and symptoms attributed to the MS, essentially in a web-like manner, resulting in the self-maintenance of most pathological traits. The increased knowledge of pathogenic paths only tends to strengthen the conclusion of the existence of a powerful linkage between the MS components.10,285,286 Nevertheless, two major issues challenge this general view: a) the lack of identification of a hierarchical line-up of the diseases, with a clearly identifiable origin for the whole MS pathology tree and branches, and b) the extreme resilience of the system to therapeutic intervention.
CONCLUSIONS
The evidence linking the function of androgens, estrogens and glucocorticoids with the MS is abundant and consistent.287
Glucocorticoids: The full development of the MS pathologies is enhanced by glucocorticoids. Glucocorticoids suppress inflammation, a central characteristic of MS, and keep the immune system at bay. However, glucocorticoids provoke insulin and leptin resistance, which aggravates the already altered energy handling control of the MS, favouring fat synthesis, protein mobilization and increased liver glucose output. Glucocorticoids also alter the ponderostat setting, thus influencing the deposition of fat both through the loss of insulin control of glycemia, increased lipogenesis and energy availability and deregulation of the ponderostat system.
Androgens, on the other hand, tend to protect body protein and maintain growth: they are probably major glucocorticoid antagonists. Patients with MS pathologies are prone to hypoandrogenism in parallel to hypercortisolism.
Estrogens are critical for control of inflammation because of their protective and antioxidant properties. They are more potent than androgens when countering the combined effects of inflammation and glucocorticoids and are thus able to maintain a more robust resistance to the MS manifestations in females than that evident in males (Table 1). At menopause, however, the differences tend to disappear because of the fall in estrogen.
In summary, the equilibrium maintained in conditions of basal health conditions between androgen, estrogen and glucocorticoids are deeply altered by the dietary and environmental causes of the MS, establishing the preponderance of glucocorticoids over the other steroid hormone types as the main factor responsible for the appearance of its related pathologies. The marked differences in MS manifestations between females and males and the increasing rates of appearance (and severity) with advancing age (and losses of androgen and estrogen) help reinforce this hypothesis.287
ACKNOWLEDGEMENTS
Supported by grant SAF2009-11739 of the Plan Nacional de Investigación en Biomedicina of the Government of Spain
REFERENCES
1.Vague J, 1956 The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr 4: 20-34.2.Hattori K, Numata N, Ikoma M, Matsuzaka A, Danielson RR, 1991 Sex differences in the distribution of subcutaneous and internal fat. Hum Biol 63: 53-63.
3.Starcke S, Vollmer G, 2006 Is there an estrogenic component in the metabolic syndrome. Genes Nutr 1: 177-188.
4.Yanase T, Fan WQ, Kyoya K, Min L, Takayanagi R, Kato S, Nawata H, 2008 Androgens and metabolic syndrome: Lessons from androgen receptor knock out (ARKO) mice. J Steroid Biochem Mol Biol 109: 254-257.
5.Cheal KL, Abbasi F, Lamendola C, McLaughlin T, Reaven GM, Ford ES, 2004 Relationship to insulin resistance of the Adult Treatment Panel III diagnostic criteria for identification of the metabolic syndrome. Diabetes 53: 1195-1200.
6.Reaven G, 2004 The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol Metabol Clin North Am 33: 283-303.
7.Maury E, Brichard SM, 2010 Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol 314: 1-16.
8.Vidal H, 2003 Obesite et inflammation: les adipocytokines. Ann Endocrinol 64: S40-S44.
9.O'Rourke RW, 2009 Molecular mechanisms of obesity and diabetes: At the intersection of weight tregulation, inflammation, and glucose homeostasis. World J Surg 33: 2007-2013.
10.Miranda PJ, DeFronzo RA, Califf RM, Guyton JR, 2005 Metabolic syndrome: Definition, pathophysiology, and mechanisms. Am Heart J 149: 33-45.
11.Potenza MV, Mechanick JI, 2009 The metabolic syndrome: Definition, global impact, and pathophysiology. Nutr Clin Pract 24: 560-577.
12.Kaplan NM, 1989 The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia and hypertension. Arch Intern Med 149: 1514-1520.
13.Bosello O, Zamboni M, 2000 Visceral obesity and metabolic syndrome. Obes Rev 1: 47-56.
14.Hayes L, Pearce MS, Firbank MJ, Walker M, Taylor R, Unwin NC, 2010 Do obese but metabolically normal women differ in intra-abdominal fat and physical activity levels from those with the expected metabolic abnormalities? A cross-sectional study. BMC Publ Hlth 10: 723.
15.Messier V, Karelis AD, Prud'homme D, Primeau V, Brochu M, Rabasa-Lhoret R, 2010 Identifying metabolically healthy but obese individuals in sedentary postmenopausal women. Obesity 18: 911-917.
16.Pascot A, Despres JP, Lemieux I, et al, 2000 Contribution of visceral obesity to the deterioration of the metabolic risk profile in men with impaired glucose tolerance. Diabetologia 43: 1126-1135.
17.Stefan N, Kantartzis K, Machann J, et al, 2008 Identification and characterization of metabolically benign obesity in humans. Arch Intern Med 168: 1609-1616.
18.Esposito K, Nicoletti G, Giugliano D, 2002 Obesity, cytokines and endothelial dysfunction: A link for the raised cardiovascular risk associated with visceral obesity. J Endocrinol Invest 25: 646-649.
19.Karelis AD, 2008 Metabolically healthy but obese individuals. Lancet 372: 1281-1283.
20.Marques-Vidal P, Pecoud A, Hayoz D, et al, 2010 Normal weight obesity: Relationship with lipids, glycaemic status, liver enzymes and inflammation. Nutr Metab Cardiovasc Dis 20: 669-675.
21.Romero-Corral A, Somers VK, Sierra-Johnson J, et al, 2010 Normal weight obesity: a risk factor for cardiometabolic dysregulation and cardiovascular mortality. Eur Heart J 31: 737-746.
22.Erlingsson S, Herard S, Leinhard OD, et al, 2009 Men develop more intraabdominal obesity and signs of the metabolic syndrome after hyperalimentation than women. Metabolism 58: 995-1001.
23.Catalano KJ, Stefanovski D, Bergman RN, 2010 Critical role of the mesenteric depot versus other intra-abdominal adipose depots in the development of insulin resistance in young rats. Diabetes 59: 1416-1423.
24.Chapados N, Collin P, Imbeault P, Corriveau P, Lavoie JM, 2008 Exercise training decreases in vitro stimulated lipolysis in a visceral (mesenteric) but not in the retroperitoneal fat depot of high-fat-fed rats. Br J Nutr 100: 518-525.
25.Ebal E, Cavalie H, Michaux O, Lac G, 2008 Visceral fat and total body fat mass correlate differently with hormones in rat. Pathol Biol 56: 283-285.
26.Marin P, Bjorntorp P, 1993 Endocrine metabolic pattern and adipose tissue distribution. Horm Res 39: 81-85.
27.Gower BA, Munoz J, Desmond R, Hilario-Hailey T, Jiao XX, 2006 Changes in intra-abdominal fat in early postmenopausal women: Effecs of hormone use. Obesity 14: 1046-1055.
28.Bryzgalova G, Lundholm L, Portwood N, et al, 2008 Mechanisms of antidiabetogenic and body weight-lowering effects of estrogen in high-fat diet-fed mice. Am J Physiol 295: E904-E912.
29.Pelleymounter MA, Baker MB, McCaleb M, 1999 Does estradiol mediate leptin's effects on adiposity and body weight? Am J Physiol 276: E955-E963.
30.Bjorntorp P, 1987 Classification of obese patients and complications related to the distribution of surplus fat. Am J Clin Nutr 45: 1120-1125.
31.Vague J, 1947 La differenciation sexuelle facteur determinant des formes de l'obesite. Presse Med 55: 339-340.
32.Tremollieres FA, Pouilles JM, Ribot CA, 1996 Relative influence of age and menopause on total and regional body composition changes in postmenopausal women. Am J Obstet Gynecol 175: 1594-1600.
33.Casson PR, Elkind-Hirsch KE, Buster JE, Homsby PJ, Carson SA, Snabes MC, 1997 Effect of postmenopausal estrogen replacement on circulating androgens. Obstet Gynecol 90: 995-998.
34.Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C, 2004 Androgens and bone. Endocr Rev 25: 389-425.
35.Tracz MJ, Sideras K, Bolona ER, et al, Clinical review: Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab 91: 2011-2016.
36.Valsamakis G, Chetty R, Anwar A, Banerjee AK, Barnett A, Kumar S, 2004 Association of simple anthropometric measures of obesity with visceral fat and the metabolic syndrome in male Caucasian and Indo-Asian subjects. Diabet Med 21: 1339-1345.
37.Rebuffe-Scrive M, Marin P, Bjorntorp P, 1991 Effect of testosterone on abdominal adipose tissue in men. Int J Obesity 15: 791-795.
38.Blouin K, Richard C, Belanger C, et al, 2003 Local androgen inactivation in abdominal visceral adipose tissue. J Clin Endocrinol Metab 88: 5944-5950.
39.Blouin K, Richard C, Brochu G, et al, 2006 Androgen inactivation and steroid-converting enzyme expression in abdominal adipose tissue in men. J Endocrinol 191: 637-649.
40.Kapoor D, Goodwin E, Channer KS, Jones TH, 2006 Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur J Endocrinol 154: 899-906.
41.Gould DC, Amoroso P, Kirby RS, 2006 Hypoandrogen-metabolic syndrome: A significant issue for men's health. BJU Int 98: 494-496.
42.Hamilton EJ, Gianatti E, Strauss BJ, et al, 2011 Increase in visceral and subcutaneous abdominal fat in men with prostate cancer treated with androgen deprivation therapy. Clin Endocrinol 74: 377-383.
43.Akishita M, Fukai S, Hashimoto M, et al, 2010 Association of low testosterone with metabolic syndrome and its components in middle-aged Japanese men. Hypertens Res 33: 587-591.
44.Tchernof A, Labrie F, 2004 Dehydroepiandrosterone, obesity and cardiovascular disease risk: a review of human studies. Eur J Endocrinol 151: 1-14.
45.Giagulli VA, Kaufman JM, Vermeulen A, 1994 Pathogenesis of the Decreased Androgen Levels in Obese Men. J Clin Endocrinol Metab 79: 997-1000.
46.Lima N, Cavaliere H, Knobel M, Halpern A, Medeiros-Neto G, 2000 Decreased androgen levels in massively obese men may be associated with impaired fuction of the gonadostat. Int J Obesity 24: 1433-1437.
47.Kupelian V, Page ST, Araujo AB, Travison TG, Bremner WJ, McKinlay JB, 2006 Low sex hormone-binding globulin, total testosterone, and symptomatic androgen deficiency are associated with development of the metabolic syndrome in nonobese men. J Clin Endocrinol Metab 91: 843-850.
48.Li CY, Giles WH, Ford ES, Liu SM, Li BY, 2010 Association of testosterone and sex hormone-binding globulin with metabolic syndrome and insulin resistance in men. Diabet Care 33: 1618-1624.
49.Matsumoto AM, Bremner WJ, 2004 Serum testosterone assays. Accuracy matters. J Clin Endocrinol Metab 89: 520-524.
50.Lea OA, Stoa KF, 1972 The binding of testosterone to different serum proteins: A comparative study. Journal of Steroid Biochemistry 3: 409-419.
51.Blouin K, Despres JP, Couillard C, et al, 2005 Contribution of age and declining androgen levels to features of the metabolic syndrome in men. Metabolism 54: 1034-1040.
52.Cleland WH, Simpson ER, Mendelson CR, 1985 Effects of aging and obesity on aromatase activity of human adipose cells. J Clin Endocrinol Metab 60: 174-177.
53.Brind J, Strain G, Miller L, Zumoff B, Vogelman J, Orentreich N, 1990 Obese men have elevated plasma levels of estrone sulfate. Int J Obesity 14: 483-486.
54.Cohen PG, 1999 The hypogonadal-obesity cycle: role of aromatase in modulating the testosterone-estradiol shunt - a major factor in the genesis of morbid obesity. Med Hypoth 52: 49-51.
55.Zumoff B, Miller LK, Strain GW, 2003 Reversal of the hypogonadotropic hypogonadism of obese men by administration of the aromatase inhibitor testolactone. Metabolism 52: 1126-1128.
56.Schaller GB 1964 The year of the gorilla, Chicago: University of Chicago Press.
57.Gesta S, Bluher M, Yamamoto Y, et al, 2006 Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Nat Acad Sci USA 103: 6676-6681.
58.Shen W, Punyanitya M, Silva AM, et al, 2009 Sexual dimorphism of adipose tissue distribution across the lifespan: a cross-sectional whole-body magnetic resonance imaging study. Nutr Metab 6: 17.
59.Cowell CT, Briody J, Lloyd-Jones S, Smith C, Moore B, Howman-Giles R, 1997 Fat distribution in children and adolescents - the influence of sex and hormones. Horm Res 48: 93-100.
60.Labayen I, Moreno LA, Blay MG, et al, 2006 Early programming of body composition and fat distribution in adolescents. J Nutr 136: 147-152.
61.Perfetto F, Tarquini R, Cornelissen G, et al, 2004 Circadian phase difference of leptin in android versus gynoid obesity. Peptides 25: 1297-1306.
62.Ahima RS, Dushay J, Flier SN, Prabakaran D, Flier JS, 1997 Leptin accelerates the onset of puberty in normal female mice. J Clin Invest 99: 391-395.
63.Ring LE, Zeltser LM, 2010 Disruption of hypothalamic leptin signaling in mice leads to early-onset obesity, but physiological adaptations in mature animals stabilize adiposity levels. J Clin Invest 120: 2931-2941.
64.Goodarzi MO, Azziz R, 2006 Diagnosis, epidemiology, and genetics of the polycystic ovary syndrome. Best Pract Res Clin Endocr Metab 20: 193-205.
65.Gilling-Smith C, Willis DS, Beard RW, Franks S, 1994 Hypersecretion of androstenedione by isolated thecal cells from polycystic ovaries. J Clin Endocrinol Metab 79: 1158-1165.
66.Ducluzeau PH, Laville M, Vidal H, Pugeat M, 2001 Resistance a l'insuline et syndrome des ovaires poly-kystiques. Diabete Metabol 27: S7-S12.
67.Orio F, Palomba S, Cascella T, et al, 2003 Adiponectin levels in women with polycystic ovary syndrome. J Clin Endocrinol Metab 88: 2619-2623.
68.Tarkun I, Arslan BC, Canturk Z, Turemen E, Sahin T, Duman C, 2004 Endothelial dysfunction in young women with polycystic ovary syndrome: Relationship with insulin resistance and low-grade chronic inflammation. J Clin Endocrinol Metab 89: 5592-5596.
69.Meyer C, McGrath BP, Teede HJ, 2005 Overweight women with polycystic ovary syndrome have evidence of subclinical cardiovascular disease. J Clin Endocrinol Metab 90: 5711-5716.
70.Pasquali R, Gambineri A, Pagotto U, 2006 The impact of obesity on reproduction in women with polycystic ovary syndrome. Br J Obstet Gynaecol 113: 1148-1159.
71.Attaoua R, El Mkadem SA, Radian S, et al, 2008 FTO gene associated to metabolic syndrome in women with polycystic ovary syndrome. Biochem Biophys Res Commun 373: 230-234.
72.Vignesh JP, Mohan V, 2007 Polycystic ovary syndrome: A component of metabolic syndrome? J Postgrad Med 53: 128-134.
73.Kumar A, Woods KS, Bartolucci AA, Azziz R, 2005 Prevalence of adrenal androgen excess in patients with the polycystic ovary syndrome (PCOS). Clin Endocrinol 62: 644-649.
74.Gilling-Smith C, Story H, Rogenrs V, Franks S, 1997 Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome. Clin Endocrinol 47: 93-99.
75.Goodarzi MO, Erickson S, Port SC, Jennrich RI, Korenman SG, 2003 Relative impact of insulin resistance and obesity on cardiovascular risk factors in polycystic ovary syndrome. Metabolism 52: 713-719.
76.Moll E, van der Veen F, van Wely M, 2007 The role of metformin in polycystic ovary syndrome: a systematic review. Hum Reprod Up 13: 527-537.
77.Essah PA, Wickham EP, Nestler JE, 2007 The metabolic syndrome in polycystic ovary syndrome. Clin Obstet Gynecol 50: 205-225.
78.Lankarani M, Valizadeh N, Heshmat R, Peimani M, Sohrabvand F, 2009 Evaluation of insulin resistance and metabolic syndrome in patients with polycystic ovary syndrome. Gynecol Endocrinol 25: 504-507.
79.Bhattacharya SM, 2010 Prevalence of metabolic syndrome in women with polycystic ovary syndrome, using two proposed definitions. Gynecol Endocrinol 26: 516-520.
80.Gould DC, Kirby RS, Amoroso P, 2007 Hypoandrogen-metabolic syndrome: a potentially common and underdiagnosed condition in men. Int J Clin Pract 61: 341-344.
81.Kaplan SA, Meehan AG, Shah A, 2006 The age related decrease in testosterone is significantly exacerbated in obese men with the metabolic syndrome. What are the implications for the relatively high incidence of erectile dysfunction observed in these men? J Urol 176: 1524-1527.
82.Corona G, Monami M, Rastrelli G, Aversa A, Tishova Y, Saad F, Lenzi A, Forti G, Mannucci E, Maggi M, 2011 Testosterone and metabolic syndrome: A meta-analysis study. J Sex Med 8: 272-283.
83.Howard JM, 2007 Common factor of cancer and the metabolic syndrome may be low DHEA. Ann Epidemiol 17: 270.
84.Charlton M, Angulo P, Chalasani N, et al, 2008 Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatol 47: 484-492.
85.Korhonen S, Hippelainen M, Vanhala M, Heinonen S, Niskanen L, 2003 The androgenic sex hormone profile is an essential feature of metabolic syndrome in premenopausal women: a controlled community-based study. Fertil Steril 79: 1327-1334.
86.Rodriguez A, Muller DC, Metter EJ, et al, 2007 Aging, androgens, and the metabolic syndrome in a longitudinal study of aging. J Clin Endocrinol Metab 92: 3568-3572.
87.Corbould A, 2008 Effects of androgens on insulin action in women: is androgen excess a component of female metabolic syndrome? Diab Metab Res Rev 24: 520-532.
88.Alexandre C, 2005 Androgens and bone metabolism. Joint Bone Spine 72: 202-206.
89.Mauras N, 2006 Growth hormone and testosterone: Effects on whole body metabolism and skeletal muscle in adolescence. Horm Res 66: 42-48.
90.Ophoff J, van Proeyen K, Callewaert F, et al, 2009 Androgen signaling in myocytes contributes to the maintenance of muscle mass and fiber type regulation but not to muscle strength or fatigue. Endocrinology 150: 3558-3566.
91.Ruzzin J, Wagman AS, Jensen J, 2005 Glucocorticoid-induced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 48: 2119-2130.
92.Natsui K, Tanaka K, Suda M, et al, 2006 High-dose glucocorticoid treatment induces rapid loss of trabecular bone mineral density and lean body mass. Osteoporosis Int 17: 105-108.
93.Schakman O, Gilson H, Kalista S, Thissen JP, 2009 Mechanisms of muscle atrophy induced by glucocorticoids. Horm Res 72: 36-41.
94.Shamlian T, Cole MG, 2006 Androgen treatment of depressive symptoms in older men: A systematic review of feasibility and effectiveness. Can J Psychiat 51: 295-299.
95.Zarrouf FA, Artz S, Griffith J, Sirbu C, Kommor M, 2009 Testosterone and depression: Systematic review and meta-analysis. J Psychiatr Pract 15: 289-305.
96.Giltay EJ, Tishova YA, Mskhalaya GJ, Gooren LJG, Saad F, Kalinchenko SY, 2010 Effects of testosterone supplementation on depressive symptoms and sexual dysfunction in hypogonadal men with the metabolic syndrome. J Sex Med 7: 2572-2582.
97.Kalynchuk LE, Gregus A, Boudreau D, Perrot-Sinal TS, 2004 Corticosterone increases depression-like behavior, with some effects on predator odor-induced defensive behavior, in male and female rats. Behav Neurosci 118: 1365-1377.
98.Burke HM, Davis MC, Otte C, Mohr DC, 2005 Depression and cortisol responses to psychological stress: A meta-analysis. Psychoneuroendocrinology 30: 846-856.
99.Zhao Y, Ma R, Shen J, Su H, Xing DM, Du LJ, 2008 A mouse model of depression induced by repeated corticosterone injections. Eur J Pharmacol 581: 113-120.
100.Killinger DW, Strutt BJ, Roncari DA, Khalil MW, 1995 Estrone formation from dehydroepiandrosterone in cultured human breast adipose stromal cells. J Steroid Biochem Mol Biol 52: 195-201.
101.Schwarz HP, 1990 Conversion of dehydroepiandrosterones sulfate (DHEA-S) to estrogens and testosterone in young nonpregnant women. Horm Metabol Res 22: 309-310.
102.Favre J, Gao J, Henry JP, et al, 2010 Endothelial estrogen receptor a plays an essential role in the coronary and myocardial protective effects of estradiol in ischemia/reperfusion. Arterioscler Thromb Vasc Biol 30: 2562-2567.
103.Xu Y, Armstrong SJ, Arenas IA, Pehowich DJ, Davidge ST, 2004 Cardioprotection by chronic estrogen or superoxide dismutase mimetic treatment in the aged female rat. Am J Physiol 287: H165-H171.
104.Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P, 2009 Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology 150: 2109-2117.
105.Windler EE, Kovanen PT, Chao YS, Brown MS, Havel RJ, Goldstein JL, 1980 The estradiol-stimulated lipoprotein receptor of rat liver. A binding site that membrane mediates the uptake of rat lipoproteins containing apoproteins B and E. J Biol Chem 255: 10464-10471.
106.Hay RV, Pottenger LA, Reingold AL, Getz GS, Wissler RW, 1971 Degradation of I125-labelled serum low density lipoprotein in normal and estrogen-treated male rats. Biochem Biophys Res Commun 44: 1471-1477.
107.Simpkins JW, Dykens JA, 2008 Mitochondrial mechanisms of estrogen neuroprotection. Brain Res Rev 57: 421-430.
108.Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM, 2009 Estradiol rescues neurons from global ischemia-induced cell death: Multiple cellular pathways of neuroprotection. Steroids 74: 555-561.
109.Guo XM, Lu X, Ren HM, Levin ER, Kassab GS, 2006 Estrogen modulates the mechanical homeostasis of mouse arterial vessels through nitric oxide. Am J Physiol 290: H1788-H1797.
110.Stirone C, Duckles SP, Krause DN, Procaccio V, 2005 Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol 68: 959-965.
111.Villa P, Sagnella F, Perri C, et al, 2008 Low- and standard-estrogen dosage in oral therapy: dose-dependent effects on insulin and lipid metabolism in healthy postmenopausal women. Climacteric 11:498-508.
112.Yoon M, 2009 The role of PPARa in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARa actions. Pharmacol Res 60: 151-159.
113.Al-Azzawi F, Palacios S, 2009 Hormonal changes during menopause. Maturitas 63: 135-137.
114.Sarrel PM, 2002 Androgen deficiency: menopause and estrogen-related factors. Fertil Steril 77: 63-67.
115.Bachmann GA, 2002 The hypoandrogenic woman: pathophysiologic overview. Fertil Steril 77: 72-76.
116.Tang V, Torregrosa C, Remesar X, Alemany M, 2001 Dietary oleoyl-estrone affects the growth rate of young rats. Eur J Nutr 40: 17-22.
117.van den Belt K, Berckmans P, Vangenechten C, Verheyen R, Witters H, 2004 Comparative study on the in vitro in vivo estrogenic potencies of 17 beta-estradiol, estrone, 17 alpha-ethynylestradiol and nonylphenol. Aquat Toxicol 66: 183-195.
118.Schlindler AE, Ebert A, Friedrich E, 1972 Conversion of androstenedione to estrone by human fat tissue. J Clin Endocrinol Metab 35: 627-630.
119.Remesar X, Fernandez-Lopez JA, Savall P, et al, 2002 Effect of oral oleoyl-estrone on adipose tissue composition in male rats. Int J Obesity 26: 1092-1102.
120.Esteve M, Virgili J, Aguilar H, et al, 1999 Leptin enhances the synthesis of oleoyl-estrone from estrone in white adipose tissue. Eur J Nutr 38: 99-104.
121.Vila R, Cabot C, Villarreal L, Monegal A, et al, 2011 Oleoyl-estrone is a precursor of an estrone-derived ponderostat signal. J Steroid Biochem Mol Biol 124: 99-111.
122.Levin BE, Patterson CM, 2005 Exercising the obese brain: Resetting the defended body weight. Endocrinology 146: 1674-1675.
123.Grasa MD, Cabot C, Esteve M, et al, 2001 Daily oral oleoyl-estrone gavage induces a dose-dependent loss of fat in Wistar rats. Obes Res 9: 202-209.
124.Buscemi S, Caimi G, Verga S, 1996 Resting metabolic rate and postabsorptive substrate oxidation in morbidly obese subjects before and after massive weight loss. Int J Obesity 20: 41-46.
125.Cottam DR, Mattar SG, Barinas-Mitchell E, et al, 2004 The chronic inflammatory hypothesis for the morbidity associated with morbid obesity: Implications and effects of weight loss. Obes Surg 14: 589-600.
126.Sola E, Jover A, Lopez-Ruiz A, et al, 2009 Parameters of inflammation in morbid obesity: lack of effect of moderate weight loss. Obes Surg 19: 571-576.
127.Levin BE, Dunn-Meynell AA, 2002 Defense of body weight depends on dietary composition and palatability in rats with diet-induced obesity. Am J Physiol 282: R46-R54.
128.Weigensberg MJ, Toledo-Corral CM, Goran MI, 2008 Association between the metabolic syndrome and serum cortisol in overweight Latino youth. J Clin Endocrinol Metab 93: 1372-1378.
129.Anagnostis P, Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP, 2009 The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J Clin Endocrinol Metab 94: 2692-2701.
130.Vogelzangs N, Beekman ATF, Dik MG, et al, 2009 Late-life depression, cortisol, and the metabolic syndrome. Am J Geriat Psychiat 17: 716-721.
131.van Steensel B, Jenster G, Damm K, Brinkmann AO, van Driel R, 1995 Domains of the human androgen receptor and glucocorticoid receptor involved in binding to the nuclear matrix. J Cell Biochem 57: 465-478.
132.Pasquali R, Vicennati V, Gambineri A, Pagotto U, 2008 Sex-dependent role of glucocorticoids and androgens in the pathophysiology of human obesity. Int J Obesity 32: 1764-1779.
133.Cheng MY, Sun G, Jin M, Zhao H, Steinberg GK, Sapolsky RM, 2009 Blocking glucocorticoid and enhancing estrogenic genomic signaling protects against cerebral ischemia. J Cereb Blood Flow Metab 29: 130-136.
134.Weiser MJ, Handa RJ, 2009 Estrogen impairs glucocorticoid dependent negative feedback on the hypothalamic-pituitary-adrenal axis via estrogen receptor alpha within the hypothalamus. Neuroscince 159: 883-895.
135.Grasa MM, Serrano M, Fernandez-Lopez JA, Alemany M, 2007 Corticosterone inhibits the lipid-mobilizing effects of oleoyl-estrone in adrenalectomized rats. Endocrinology 148: 4056-4063.
136.Wolf G, 2002 Glucocorticoids in adipocytes stimulate visceral obesity. Nutr Rev 60: 148-151.
137.Wang M, 2005 The role of glucocorticoid action in the pathophysiology of the metabolic syndrome. Nutr Metab 2: 3.
138.Macfarlane DP, Forbes S, Walker BR, 2008 Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome. J Endocrinol 197: 189-204.
139.Napolitano A, Voice MW, Edwards CRW, Seckl JR, Chapman KE, 1998 11b-hydroxysteroid dehydrogenase 1 in adipocytes: Expression is differentiation-dependent and hormonally regulated. J Steroid Biochem Mol Biol 64: 251-260.
140.Tomlinson JW, Stewart PM, 2002 The functional consequences of 11b-hydroxysteroid dehydrogenase expression in adipose tissue. Horm Metabol Res 34: 746-751.
141.Lee MJ, Fried SK, Mundt SS, Wang Y, Sullivan S, Stefanni A, Daugherty BL, Hermanowski-Vosatka A, 2008 Depot-specific regulation of the conversion of cortisone to cortisol in human adipose tissue. Obesity 16: 1178-1185.
142.Stimson RH, Andersson J, Andrew R, et al, 2009 Cortisol release from adipose tissue by 11b-Hydroxysteroid Dehydrogenase type 1 in humans. Diabetes 58: 46-53.
143.Muller J, 1995 Aldosterone: The minority hormone of the adrenal cortex. Steroids 60: 2-9.
144.Hornsby PJ, 1995 Biosynthesis of DHEAS by the human adrenal cortex and its age-related decline. Ann NY Acad Sci 774: 29-46.
145.Havelock JC, Auchus RJ, Rainey WE, 2004 The rise in adrenal androgen biosynthesis: Adrenarche. Semin Reprod Med 22: 337-347.
146.Reynolds RM, Walker BR, 2003 Human insulin resistance: the role of glucocorticoids. Diabet Obes Metabol 5: 5-12.
147.Asensio C, Muzzin P, Rohner-Jeanrenaud F, 2004 Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int J Obesity 28: S45-S52.
148.Zakrzewska KE, Cusin I, Sainsbury A, Rohner-Jeanrenaud F, Jeanrenaud B, 1997 Glucocorticoids as counterregulatory hormones of leptin - Toward an understanding of leptin resistance. Diabetes 46: 717-719.
149.Ray DW, 1996 Molecular mechanisms of glucocorticoid resistance. J Endocrinol 149: 1-5.
150.Ray PD, Foster DO, Lardy HA, 1964 Mode of action of glucocorticoids. I. Stimulation of gluconeogenesis independent of synthesis de novo of enzymes. J Biol Chem 239: 3396-3400.
151.May RC, Bailey JL, Mitch WE, Masud T, England BK, 1996 Glucocorticoids and acidosis stimulate protein and amino acid catabolism in vivo. Kidney Int 49: 679-683.
152.Tauchmanova L, Pivonello R, di Somma C, Rossi R, de Martino MC, Camera L, Klain M, Salvatore M, Lombardi G, Colao A, 2006 Bone demineralization and vertebral fractures in endogenous cortisol excess: Role of disease etiology and gonadal status. J Clin Endocrinol Metab 91: 1779-1784.
153.Franchimont D, 2004 Overview of the actions of glucocorticoids on the immune response. A good model to characterize new pathways of immunosuppression for new treatment strategies. Ann NY Acad Sci 1024: 124-137.
154.Kirschbaum C, Prussner JC, Stone AA, et al, 1995 Persistent high cortisol responses to repeated psychological stress in a subpopulation of healthy men. Psychosomat Med 57: 468-474.
155.Duclos M, Gouarne C, Bonnemaison D, 2003 Acute and chronic effects of exercise on tissue sensitivity to glucocorticoids. J Appl Physiol 94: 869-875.
156.Jensen J, Riis BJ, Strom V, Nilas L, Christiansen C, 1987 Long-term effects of percutaneous estrogens and oral progesterone on serum lipoproteins in postmenopausal women. Am J Obstet Gynecol 156: 66-71.
157.Wortsman J, Frank S, Cryer PE, 1984 Adrenomedullary response to maximal stress in humans. Am J Med 77: 779-784.
158.Sutter-Dub MT, 2002 Rapid non-genomic and genomic responses to progestogens, estrogens, and glucocorticoids in the endocrine pancreatic B cell, the adipocyte and other cell types. Steroids 67: 77-93.
159.Kochakian CD, 1975 Definition of androgens and protein anabolic steroids. Pharmacol Ther B 1: 149-177.
160.Pasquali R, Vicennati V, Cacciari M, Pagotto U, 2006 The hypothalamic-pituitary-adrenal axis activity in obesity and the metabolic syndrome. Ann NY Acad Sci 1083: 111-128.
161.Chrousos GP, 2000 The role of stress and the hypothalamic-pituitary-adrenal axis in the pathogenesis of the metabolic syndrome: neuro-endocrine and target tissue-related causes. Int J Obesity 24: Suppl 2: 50-55.
162.Rosmond R, 2005 Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinol 30: 1-10.
163.Hautanen A, Raikkonen K, Adlerkreutz H, 1997 Associations between pituitary-adrenocortical function and abdominal obesity, hyperinsulinaemia and dyslipidaemia in normotensive males. J Intern Med 241: 451-461.
164.Livingstone DEW, Jones GC, Smith K, et al, 2000 Understanding the role of glucocorticoids in obesity: Tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology 141: 560-563.
165.Wang MH, 2006 Tissue-specific glucocorticoid excess in the metabolic syndrome: 11b-HSD1 as a therapeutic target. Drug Develop Res 67: 567-569.
166.Grasa MM, Cabot C, Fernandez-Lopez JA, Remesar X, Alemany M, 2001 Modulation of corticosterone availability to white adipose tissue of lean and obese Zucker rats by corticosteroid-binding globulin. Horm Metabol Res 33: 407-411.
167.Barat P, Duclos M, Gatta B, Roger P, Mormede P, Moisan MP, 2005 Corticosteroid binding globulin gene polymorphism influences cortisol driven fat distribution in obese women. Obes Res 13: 1485-1490.
168.Bledsoe RK, Montana VG, Stanley TB, et al, 2002 Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110: 93-105.
169.Evans RM, 1988 The steroid and thyroid hormone receptor superfamily. Science 240: 889-895.
170.Chen SY, Wang J, Yu GQ, Liu WH, Pearce D, 1997 Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. J Biol Chem 272:14087-14092.
171.Mitra R, Sapolsky RM, 2010 Expression of chimeric estrogen-glucocorticoid-receptor in the amygdala reduces anxiety. Brain Res 1342: 33-38.
172.Mackem S, Baumann CT, Hager GL, 2001 A glucocorticoid/retinoic acid receptor chimera that displays cytoplasmic/nuclear translocation in response to retinoic acid. A real time sensing assay for nuclear receptor ligands. J Biol Chem 276: 45501-45504.
173.Genaro G, Franci CR, 2010 Cortisol influence on testicular testosterone secretion in domestic cat: An in vitro study. Pesq Vet Bras 30:887-890.
174.Dong Q, Salva A, Sottas CM, Niu EM, Holmes M, Hardy MR, 2004 Rapid glucocorticoid mediation of suppressed testosterone biosynthesis in male mice subjected to immobilization stress. J Androl 25: 973-981.
175.MacAdams MR, White RH, Chipps BE, 1986 Reduction of serum testosterone levels during chronic glucocorticoid therapy. Ann Int Med 104: 648-651.
176.Labrie F, Belanger A, Simard J, Luu-The V, Labrie C, 1995 DHEA and peripheral androgen and estrogen formation: intracrinology. Ann NY Acad Sci 774: 16-28.
177.Apostolova G, Schweizer RAS, Balazs Z, Kostadinova RM, Odermatt A, 2005 Dehydroepiandrosterone inhibits the amplification of glucocorticoid action in adipose tissue. Am J Physiol 288: E957-E964.
178.Browne ES, Wright BE, Porter JR, Svec F, 1992 Dehydroepiandrosterone. Antiglucocorticoid action in mice. Am J Med Sci 303: 366-371.
179.Gong H, Jarzynka MJ, Cole TJ, et al, 2008 Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res 68: 7386-7393.
180.Weiser MJ, Foradori CD, Handa RJ, 2010 Estrogen receptor beta activation prevents glucocorticoid receptor-dependent effects of the central nucleus of the amygdala on behavior and neuroendocrine function. Brain Res 1336: 78-88.
181.Simoncini T, Maffei S, Basta G, et al, 2000 Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circul Res 87: 19-25.
182.da Silva JP, 1999 Sex hormones and glucocorticoids: Interactions with the immune system. Ann NY Acad Sci 876: 102-118.
183.Sheffield-Moore M, 2000 Androgens and the control of skeletal muscle protein synthesis. Ann Med 32: 181-186.
184.Isozaki Y, Mitch WE, England BK, Price SR, 1996 Protein degradation and increased mRNAs encoding proteins of the ubiquitin-proteasome proteolytic pathway in BC3H1 myocytes require an interaction between glucocorticoids and acidification. Proc Nat Acad Sci USA 93: 1967-1971.
185.Burt MG, Gibney J, Ho KKY, 2007 Protein metabolism in glucocorticoid excess: study in Cushing's syndrome and the effect of treatment. Am J Physiol 292: E1426-E1432.
186.Goldstein RE, Rossetti L, Palmer BA, et al, 2002 Effects of fasting and glucocorticoids on hepatic gluconeogenesis assessed using two independent methods in vivo. Am J Physiol 283: E946-E957.
187.Monroe SE, Menon KMJ, 1977 Changes in reproductive hormone secretion during the climacteric and postmenopausal periods. Clin Obstet Gynecol 20: 113-122.
188.Toth MJ, Tchernof A, Sites CK, Poehlman ET, 2000 Effect of menopausal status on body composition and abdominal fat distribution. Int J Obesity 24: 226-231.
189.Franklin RM, Ploutz-Snyder L, Kanaley JA, 2009 Longitudinal changes in abdominal fat distribution with menopause. Metabolism 58: 311-315.
190.Macdonald HM, New SA, Campbell MK, Reid DM, 2003 Longitudinal changes in weight in perimenopausal and early postmenopausal women: effects of dietary energy intake, energy expenditure, dietary calcium intake and hormone replacement therapy. Int J Obesity 27: 669-676.
191.Piche ME, Weisnagel SJ, Corneau L, Nadeau A, Bergeron J, Lemieux S, 2005 Contribution of abdominal visceral obesity and insulin resistance to the cardiovascular risk profile of postmenopausal women. Diabetes 54: 770-777.
192.Motivala AA, Rose PA, Kim HM, et al, 2008 Cardiovascular risk, obesity, and myocardial blood flow in postmenopausal women. J Nucl Cardiol 15: 510-517.
193.Mesch VR, Siseles NO, Maidana PN, et al, 2008 Androgens in relationship to cardiovascular risk factors in the menopausal transition. Climacteric 11: 509-517.
194.Korytkowski MT, Krug EI, Daly MA, DeRiso L, Wilson JW, Winters SJ, 2005 Does androgen excess contribute to the cardiovascular risk profile in postmenopausal women with type 2 diabetes? Metabolism 54: 1626-1631.
195.Bonomo SM, Rigamonti AE, Giunta M, Galimberti D, Guaita A, Gagliano MG, Muller EE, Cella SG, 2009 Menopausal transition: A possible risk factor for brain pathologic events. Neurobiol Aging 30: 71-80.
196.Kaye SA, Folsom AR, 1991 Is serum cortisol associated with body-fat distribution in postmenopausal women. Int J Obesity 15: 437-439.
197.Lindsay R, 1996 The menopause and osteoporosis. Obstet Gynecol 87:16S-19S.
198.Tenover JL, 1967 Testosterone and the aging male. J Androl 18: 103-106.
199.Delaporte E, 2008 Affections inflammatoires a mediation immunitaire et psoriasis. Ann Dermatol 135: Suppl 4: 269-274.
200.Karadag AS, Yavuz B, Ertugrul DT, et al, 2010 Is psoriasis a pre-atherosclerotic disease? Increased insulin resistance and impaired endothelial function in patients with psoriasis. Int J Dermatol 49: 642-646.
201.Reutrakul S, Hathout EH, Janner D, et al, 2004 Familial juvenile autoimmune hypothyroidism, pituitary enlargement, obesity, and insulin resistance. Thyroid 14: 311-319.
202.Mauras N, DelGiorno C, Kollman C, et al, 2010 Obesity without established comorbidities of the metabolic syndrome is associated with a proinflammatory and prothrombotic state, even before the onset of puberty in children. J Clin Endocrinol Metab 95: 1060-1068.
203.van Dijk SJ, Feskens EJM, Bos MB, et al, 2009 A saturated fatty acid-rich diet induces an obesity-linked proinflammatory gene expression profile in adipose tissue of subjects at risk of metabolic syndrome. Am J Clin Nutr 90: 1656-1664.
204.Fleming ME, Sales KM, Winslet MC, 2005 Diet and colorectal cancer: implications for the obese and devotees of the Atkins diet. Colorect Dis 7: 128-132.
205.Jaggers JR, Sui XM, Hooker P, et al, 2009 Metabolic syndrome and risk of cancer mortality in men. Eur J Cancer 45: 1831-1838.
206.Russo A, Autelitano M, Bisanti L, 2008 Metabolic syndrome and cancer risk. Eur J Cancer 44: 293-297.
207.Tubiash HS, 1951 The anaerogenic effect of nitrates and nitrites on Gram-negative enteric bacteria. Am J Publ Hlth 41: 833-838.
208.Dunca BB, Schmidt MI, 2001 Chronic activation of the innate immune system may underlie the metabolic syndrome. S Paulo Med J 119: 122-127.
209.Cani PD, Possemiers S, van de Wiele T, et al, 2009 Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58: 1091-1103.
210.Ley RE, Turnbaugh PJ, Klein S, Gordon JI, 2006 Human gut microbes associated with obesity. Nature 444: 1022-1023.
211.Najzer M, Seeley RJ, 2006 Obesity and gut flora. Nature 444: 1009-1010.
212.Delzenne NM, Cani PD, 2010 Nutritional modulation of gut microbiota in the context of obesity and insulin resistance: Potential interest of prebiotics. Int Dairy J 20: 277-280.
213.Manco M, Putignani L, Bottazzo GF, 2010 Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev 31: 817-844.
214.Creely SJ, McTernan PG, Kusminski CM, et al, 2007 Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol 292: E740-E747.
215.Boivin MA, Ye D, Kennedy JC, Al-Sadi R, Shepela C, Ma TY, 2007 Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am J Physiol 292: G590-G598.
216.Hales CN, Barker DJP, 2001 The thrifty phenotype hypothesis. Br Med Bull 60: 5-20.
217.Flodmark CE, 2002 Thrifty genotypes and phenotypes in the pathogenesis of early-onset obesity. Acta Paediatr 91: 737-738.
218.Sclafani A, Springer D, 1976 Dietary obesity in adult rats: Similarities to hypothalamic and human obesity syndromes. Physiol Behav 17: 461-471.
219.Prats E, Monfar M, Iglesias R, Castella J, Alemany M, 1989 Energy intake of rats fed a cafeteria diet. Physiol Behav 45: 263-272.
220.Rogers PJ, 1985 Returning 'cafeteria-fed' rats to a chow diet: negative contrast and effects of obesity on feeding behaviour. Physiol Behav 35: 493-499.
221.Welle SL, Amatruda JM, Forbes GB, Lockwood DH, 1984 Resting metabolic rates of obese women after rapid weight loss. J Clin Endocrinol Metab 59: 113-122.
222.Brownell KD, Greenwood MRC, Stellar E, Shrager EE, 1986 The effects of repeated cycles of weight loss and regain in rats. Physiol Behav 38: 459-464.
223.Kirschner MA, Schneider G, Ertel NH, Gorman J, 1988 An 8-year experience with a very-low-calorie formula diet for control of major obesity. Int J Obesity 12: 69-80.
224.van Itallie TB, Kral JG, 1981 The dilemma of morbid obesity. J Am Med Assoc 246: 999-1003.
225.Dulloo AG, Girardier L, 1992 Influence of dietary composition on energy expenditure during recovery of body weight in the rat. Implications for catch up growth and obesity relapse. Metabolism 41: 1336-1342.
226.Bradley P, 1978 The ponderostat and a physiological model of obesity. Am J Clin Nutr 31: 1976-1978.
227.Dube MG, Xu B, Crowley WR, Kalra PS, Kalra SP, 1994 Evidence that neuropeptide Y is a physiological signal for normal food intake. Brain Res 646: 341-344.
228.Yang L, Scott KA, Hyun J, et al, 2009 Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and energy balance. J Neurosci 29: 179-190.
229.Benoit S, Schwartz M, Baskin D, Woods SC, Seeley RJ, 2000 CNS melanocortin system involvement in the regulation of food intake. Horm Behav 37: 299-305.
230.Ellacott KLJ, Cone RD, 2006 The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Phil Trans Roy Soc Lond B 361: 1265-1274.
231.Raffin-Sanson ML, de Keyzer Y, Bertagna X, 2003 Proopiomelanocortin, a polypeptide precursor with multiple functions: from physiology to pathological conditions. Eur J Endocrinol 149: 79-90.
232.Costin GE, Hearing VJ, 2007 Human skin pigmentation: melanocytes modulate skin color in response to stress. FASEB J 21: 976-994.
233.Schwartz RA, 1994 Acanthosis nigricans. J Am Acad Dermatol 31: 1-19.
234.Kahn CR, Flier JS, Bar RS, et al, 1976 The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N Engl J Med 294: 739-745.
235.Lopez M, Lage R, Tung CL, et al, 2007 Orexin expression is regulated by alpha-melanocyte stimulating hormone. J Neuroendocrinol 19: 703-707.
236.Rodgers RJ, Halford JCG, Nunes de Souza RL, et al, 2000 Dose-response effects of orexin-A on food intake and the behavioural satiety sequence in rats. Regul Peptides 96: 71-84.
237.Salin-Pascual RI, 2001 The role of the hypothalamic neuropeptides hypocretin/orexin in the sleep-wake cycle. Isr Med Assoc J 3: 144-146.
238.Bass J, Turek FW, 2005 Sleepless in America. A pathway to obesity and the metabolic syndrome? Arch Intern Med 165: 15-16.
239.Choi KM, Lee JS, Park HS, Baik SH, Choi DS, Kim SM, 2008 Relationship between sleep duration and the metabolic syndrome:Korean National Health Nutrition Survey 2001. Int J Obesity 32: 1091-1097.
240.Donga E, van Dijk M, van Dijk JG, et al, 2010 A single night of partial sleep deprivation induces insulin resistance in multiple metabolic pathways in healthy subjects. J Clin Endocrinol Metab 95: 2963-2968.
241.Rosa Neto JC, Lira FS, Venancio DP, et al, 2010 Sleep deprivation affects inflammatory marker expression in adipose tissue. Lip Hlth Dis 9: 125.
242.Hasler G, Buysse DJ, Klaghofer R, et al, 2004 The association between short sleep duration and obesity in young adults: A 13-year prospective study. Sleep 27: 661-666.
243.Nielsen LS, Danielsen KV, Sorensen TIA, 2011 Short sleep duration as a possible cause of obesity: critical analysis of the epidemiological evidence. Obes Rev 12: 78-92.
244.Resta O, Barbaro MPF, Bonfitto P, et al, 2003 Low sleep quality and daytime sleepiness in obese patients without obstructive sleep apnoea syndrome. J Intern Med 253: 536-543.
245.Fukagawa K, Sakata T, Yoshimatsu H, Fujimoto K, Uchimura K, Asano C, 1992 Advance shift of feeding circadian rhythm induced by obesity progression in Zucker rats. Am J Physiol 263: R1169-R1175.
246.Dickmeis T, Lahiri K, Nica G, et al, 2007 Glucocorticoids play a key role in circadian cell cycle rhythms. PLoS Biol 5: 854-864.
247.Dallman MF, Viau VG, Bhatnagar S, Gomez F, Laugero K, Beli ME, 2011 Corticotropin-releasing factor, corticosteroids, stress, and sugar: energy balance, the brain and behavior. Hormones Brain and Behavior 1: 571-631.
248.Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB, 2005 Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol 493: 63-71.
249.Lopez M, Varela L, Vazquez MJ, et al, 2010 Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med 16: 1001-1008.
250.Handa RJ, Burgess LH, Kerr JE, O'Keefe JA, 1994 Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm Behav 28: 464-476.
251.Kiss A, Jezova D, Aguilera G, 1994 Activity of the hypothalamic-pituitary-adrenal axis and sympathoadrenal system during food and water deprivation in the rat. Brain Res 663: 84-92.
252.Yoshimatsu H, Niijima A, Oomura Y, Katafuchi T, 1988 Lateral and ventromedial hypothalamic influences on hepatic autonomic nerve activity in the rat. Brain Res Bull 21: 239-244.
253.Atrens DM, Holmes LJ, Jirasek M, Siviy SM, Solowij N, 1987 Hypothalamic modulation of thermogenesis and energy substrate utilization. Brain Res Bull 18: 303-308.
254.Kuo JJ, Silva AA, Hall JE, 2003 Hypothalamic melanocortin receptors and chronic regulation of arterial pressure and renal function. Hypertension 41: 768-774.
255.Wise RA, 2006 Role of brain dopamine in food reward and reinforcement. Phil Trans Roy Soc Lond B 361: 1149-1158.
256.Meguid MM, Fetissov SO, Varma M, et al, 2000 Hypothalamic dopamine and serotonin in the regulation of food intake. Nutrition 16: 843-857.
257.Kelley AE, Baldo BA, Pratt WE, Will MJ, 2005 Corticostriatal-hypothalamic circuitry and food motivation: Integration of energy, action and reward. Physiol Behav 86: 773-795.
258.Grill HJ, 2006 Distributed neural control of energy balance: Contributions from hindbrain and hypothalamus. Obesity 14: Suppl 5: 216-221.
259.Cummings DE, Overduin J, 2007 Gastrointestinal regulation of food intake. J Clin Invest 117: 13-23.
260.Colombani AL, Carneiro L, Benani A, et al, 2009 Enhanced hypothalamic glucose sensing in obesity: Alteration of redox signaling. Diabetes 58: 2189-2197.
261.Horn EM, Waldrop TG, 1997 Oxygen-sensing neurons in the caudal hypothalamus and their role in cardiorespiratory control. Resp Physiol 110: 219-228.
262.Mason WT, 1980 Supraoptic neurones of rat hypothalamus are osmosensitive. Nature 287: 154-157.
263.Rothwell NJ, Stock MJ, 1987 Effect of diet and fenfluramine on thermogenesis in the rat: Possible involvement of serotonergic mechanisms. Int J Obesity 11: 319-324.
264.Heal DJ, Cheetham SC, Prow MR, Martin KF, Buckett WR, 1998 A comparison of the effects on central 5-HT function of sibutramine hydrochloride and other weight-modifying agents. Br J Pharmacol 125: 301-308.
265.Levine LR, Rosenblatt S, Bosomworth J, 1987 Use of a serotonin re-uptake inhibitor, fluoxetine, in the treatment of obesity. Int J Obesity 11: 185-190.
266.Nielsen JA, Chapin DS, Johnson JL, Torgersen LK, 1992 Sertraline, a serotonin uptake inhibitor, reduces food intake and body weight in lean rats and genetically obese mice. Am J Clin Nutr 55:Suppl 1: 185-188.
267.Yu S, Holsboer F, Almeida OFX, 2008 Neuronal actions of glucocorticoids: Focus on depression. J Steroid Biochem Mol Biol 108: 300-309.
268.Porter RJ, Gallagher P, Watson S, Young AH, 2004 Corticosteroid-serotonin interactions in depression: a review of the human evidence. Psychopharmacol 173: 1-17.
269.Gomez R, Navarro M, Ferrer B, et al, 2002 A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci 22: 9612-9617.
270.Boyd ST, Fremming BA, 2005 Rimonabant - A selective CB1 antagonist. Ann Pharmacother 39: 684-690.
271.Caul WF, Jones JR, Barrett RJ, 1988 Amphetamine's effects on food consumption and body weight. The role of adaptive processes. Behav Neurosci 102: 441-450.
272.Bukowiecki L, Follea N, Jahjah L, 1982 Ephedrine, a potential slimming drug, directly stimulates thermogenesis in brown adipocytes via β-adrenoreceptors. Int J Obesity 6: 343-350.
273.Michel C, Cabanac M, 1999 Effects of dexamethasone on the body weight set point of rats. Physiol Behav 68: 145-150.
274.Messerli FH, Bell DSH, Fonseca V, et al, 2007 Body weight changes with beta-blocker use: Results from GEMINI. Am J Med 120: 610-615.
275.Patten SB, Williams JVA, Lavorato DH, Brown L, McLaren L, Eliasziw M, 2009 Major depression, antidepressant medication and the risk of obesity. Psychoter Psychosomat 78: 182-186.
276.Vanina Y, Podolskaya A, Sedky K, et al, 2002 Body weight changes associated with psychopharmacology. Psychiat Serv 53: 842-847.
277.Fornari A, Pedrazzi P, Lippi G, Picciotto MR, Zoli M, Zini I, 2007 Nicotine withdrawal increases body weight, neuropeptide Y and Agouti-related protein expression in the hypothalamus and decreases uncoupling protein-3 expression in the brown adipose tissue in high-fat fed mice. Neurosci Lett 411: 72-76.
278.Keck PE, McElroy SL, 2003 Bipolar disorder, obesity, and pharmacotherapy-associated weight gain. J Clin Psychiat 64: 1426-1435.
279.Mussell MP, Mitchell JE, de Zwaan M, Crosby RD, Seim HC, Crow SJ, 1996 Clinical characteristics associated with binge eating in obese females: A descriptive study. Int J Obesity 20: 324-331.
280.Fandino J, Moreira RO, Preissler C, et al, 2010 Impact of binge eating disorder in the psychopathological profile of obese women. Comprehens Psychiat 51: 110-114.
281.Fairburn CG, Cooper Z, Doll HA, Norman P, O'Connor M, 2000 The natural course of bulimia nervosa and binge eating disorder in young women. Arch Gen Psychiat 57: 659-665.
282.Boggiano MM, Artiga AI, Pritchett CE, Chandler-Laney PC, Smith ML, Eldridge AJ, 2007 High intake of palatable food predicts binge-eating independent of susceptibility to obesity: an animal model of lean vs obese binge-eating and obesity with and without binge-eating. Int J Obesity 31: 1357-1367.
283.Dallman MF, Pecoraro N, Akana SF, et al, 2003 Chronic stress and obesity: A new view of "comfort food". Proc Nat Acad Sci USA 100: 11696-11701.
284.Kandiah J, Yake M, Jones J, Meyer M, 2006 Stress influences appetite and comfort food preferences in college women. Nutr Res 26: 118-123.
285.Belsare PV, Watve MG, Ghaskadbi SS, Bhat DS, Yajnik CS, Jog M, 2010 Metabolic syndrome: Aggression control mechanisms gone out of control. Med Hypoth 74: 578-589.
286.Alemany M, 2011 The defense of adipose tissue against excess substrate-induced hyperthrophia: Immune system cell infiltration and arrested metabolic activity. J Clin Endocrinol Metab 96: 66-68.
287.Alemany M, 2012 Do the interactions between glucocorticoids and sex hormones regulate the development of the metabolic syndrome? Front Endocrinol 3: 27.
Address for correspondence:
Prof. Marià Alemany PhD; Department of Nutrition and Food Science, Faculty of Biology, University of Barcelona; Av. Diagonal, 643; 08028 Barcelona, Spain, Tel.: 34934034606; Fax: 34934037064; E-mail: malemany@ub.edu
Received 12-03-12, Revised 01-04-12, Accepted 30-04-12