Department of Endocrinology, the Oxford Centre for Diabetes, Endocrinology and Metabolism, Churchill Hospital, Oxford, UK
Acromegaly, a multisystemic disease resulting from excessive growth hormone (GH) and insulin-like growth factor-I (IGF-I) levels in adults, is associated with a two-to-threefold increase in mortality. The available treatment options (surgery, radiotherapy and medical treatment with somatostatin analogues or dopamine agonists) fail to achieve the currently accepted goals of therapy in a substantial number of patients. Pegvisomant, a newly developed GH receptor antagonist, represents a novel treatment modality for this disease. It binds with the GH receptor and induces internalization, but blocks receptor signaling events, thereby reducing IGF-I production. The two main published studies suggest that it is the most potent medical therapy with greater specificity, without being dependent on the tumour characteristics. However, apart from its high cost and the dilemmas raised concerning the appropriateness of using serum IGF-I concentrations as a marker of disease activity, it may also occasionally be associated with elevations in liver enzyme levels. Further studies are required to confirm its high success rates as well as to investigate the possibility of inducing an increase in the pituitary tumour size. Currently, pegvisomant is a second line treatment for acromegaly with an adjuvant role and possibly of greater value in cases of resistance to other therapeutic options.
Acromegaly, Growth hormone receptor antagonists, Pegvisomant.
INTRODUCTION
Growth hormone (GH), a protein of 191 amino acids, is synthesized and secreted by the somatotrop cells of the anterior pituitary gland. The major form found in the circulation has a molecular mass of 22 kDa. Studies with X-ray crystallography and employing a genetically engineered variant of porcine (p)GH reveal that its 3-dimensional structure consists of 4 á-helices (each ranging from 20-30 residues) joined together by non-helical polypeptide chains as well as two disulfide bonds (between Cys residues 53 and 164 and 181 and 189)1 (Figure 1). Subsequent studies on human GH demonstrated similar topographies2. Up to 60% of circulating serum GH is bound to the growth hormone binding protein (GHBP), which corresponds to part of the extracellular domain of the growth hormone receptor (GHR)3.
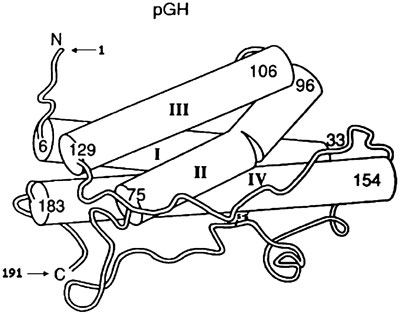
Figure 1. Crystal structure of porcin (p) GH. The four antiparallel á-helical bundles labeled with Roman numerals I_IV are demonstrated as cylindrical rods. The amino and carboxyl termini are indicated with an N and C, respectively. The numbers show amino acid positions at the N and C termini of each independent á-helix [reprinted from (3), with permission from Elsevier].
GH exerts its actions through a specific receptor belonging to the class I haematopoietin or cytokine/growth hormone/prolactin receptor superfamily. GHR is a single membrane-spanning type I glycoprotein withan extracellular ligand-binding domain, a single 24-amino acid hydrophophobic transmembrane region and an intracellular domain3 (Figure 2). It shows a wide distribution including muscle, adipose and immune tissues, liver, mammary gland, bones, kidneys, brain and embryonic stem cells4. Elegant studies in the early 1990s suggested that binding of GH to GHR resulted in receptor dimerization via a two-step mechanism2,5 involving two distinct sites (a high-affinity binding site 1, made up of amino acid residues between the C-terminal half of helix four and the N-terminal region of helix one, and a low-affinity binding site 2, made-up by N-terminal residues of the first and third helices of a single GH molecule), which bound sequentially to two GHR monomers. The subsequent dimerization lead to activation of a GHR-associated cytoplasmic tyrosine kinase, Janus-kinase 2 (JAK2), providing the initiating step of GHR signal transduction. It should be noted, though, that recent evidence supports the view that the GHR exists as a preformed dimer. GH binding to the preformed dimer occurs through sites 1 and 2 and induces a conformational change in the receptor leading to activation of JAK26 (Figure 3). The downstream signaling pathways resulting in GH actions include, but are not probably limited to, the signal transducer and activator of transcription (STAT), mitogen-activated-kinase (MAP) and phosphoinositide 3-kinase (PI3) pathways. According to the dual effector theory of GH action, its effects are mediated either directly or mainly indirectly via the production of insulin-like growth factor-I (IGF-I) by the liver, bones and other types of tissues7. Notably, IGF-I acts in an endocrine, paracrine or autocrine fashion8.
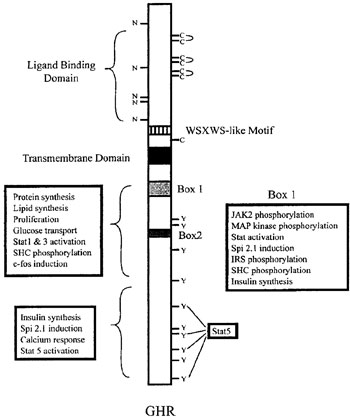
Figure 2. Structure of the GHR. Potential N-linked glycosylation sites (N) and extracellular cysteines (C) with three pairs of disulfide bonds are shown. Ten intracellular tyrosine (Y) residues important in rat GHR, and regions in the receptor required for signaling are also demonstrated [reprinted from (3), with permission from Elsevier].
THE DEVELOPMENT OF PEGVISOMANT
The development of pegvisomant represents a remarkable story taking place during attempts to produce a GH superagonist by causing mutations to the hormone molecule. Thus, it was initially observed that transgenic mice, expressing GH with mutations in amino acid residues of the 3rd helix in the region of binding site 2, exhibited a dwarf phenotype with decreased IGF-I concentrations9,10. Further studies revealed that glycine at position 120 of human GH or at position 119 of bovine GH (in the region of binding site 2) was essential for the growth promoting activity, and substitution of this with any amino acid other than alanine resulted in a GH antagonist2,11. Finally, when mutations in this region were combined with amino acid substitutions in binding site 1 (providing an increased binding affinity for the GH receptor), highly potent GH-receptor antagonists were produced12.
One such compound, the recombinant protein B2036, is produced by substitution of glycine by lysine in position 120 of binding site 2. It also differs from the wild type GH by 8 amino acids in the region of binding site 1. The latter modifications are associated with enhanced receptor affinity at site 1 and give B2036 a competitive binding advantage over the wild-type GH13. It has been demonstrated that B2036 binds to the pre-formed GHR dimers and induces internalization, but not the signalling events14 (Figure 3). B2036-PEG (Pegvisomant), a molecule with a molecular mass of 42-46 kDa, is produced when four or five polyethylene glycol (PEG)-5000 moieties are covalently attached to Lys residues of B203615. The conjugation of B2036 to polyethylene glycol (PEG)-5000 reduces its antigenicity and prolongs its half-life (from 11 min to in excess of 72 hours) by increasing its hydrodynamic volume and reducing its renal clearance16. It should be noted, however, that pegylation decreases the antagonistic activity of B2036 and, therefore, levels much higher than endogenous GH concentrations are necessary for effective blocking of GHR17. In vitro studies have shown that pegvisomant does not interact with the human prolactin receptor and, consequently, has no effects on the biological activities mediated by this receptor18.

Figure 3. Hypothesis for interaction of GH and B2036 with the GHR receptor.(1) GH binds through site 1 to GHBP. Binding to the GHR through sites 1 and 2 induces a conformational change, signalling and internalisation. (2) B2036 binds preferentially to GHBP because of the site 1 mutations (1*). However, the site 2 mutation (x) blocks the conformational change required for signalling. Internalisation nevertheless occurs [reprinted from (17), Copyright 2001, The Endocrine Society].
STUDIES WITH PEGVISOMANT IN HEALTHY VOLUNTEERS
The effects of pegvisomant on serum IGF-I and GH release were evaluated in a phase I, placebo-controlled, single rising dose study performed in 36 normal young men19. The volunteers were randomized into four groups and received a single subcutaneous injection of either placebo or pegvisomant at doses of 0.03, 0.1, 0.3 or 1.0 mg/kg. A dose-dependent increase in pegvisomant levels was observed, with peak concentrations occurring 36 h after dose administration. Serum IGF-I levels showed a dose-dependent suppression that reached statistical significance on day 3 after administration of 0.3 mg/kg. The decrease was most prominent on day 5 in response to 1.0 mg/kg of pegvisomant (49±6%), whereas serum GH levels measured by two independent methods did not change substantially. All doses of pegvisomant were well tolerated without causing serious adverse reactions.
In a later study aiming mainly to explore the feedback role of peripheral IGF-I signalling, it was demonstrated that the acute subcutaneous administration of pegvisomant in healthy eumetabolic adults (1mg/kg) suppressed total plasma IGF-I concentration by 31% 84 hours after the drug injection. It should be noted, however, that this fall was associated with elevation of the 10-hour mean serum GH concentration by 71% via augmentation of both GH secretory burst amplitude (2.1-fold) and basal/non-pulsatile GH release (2.5-fold)20.
Interestingly, it has also been shown that, in contrast to octreotide, pegvisomant administration in healthy male subjects is not associated with deterioration of glucose tolerance as a result of impaired insulin release, or alterations in gastrin, PP and CCK responses to glucose or a standard mixed meal. These findings provide good evidence for the hypothesis that pegvisomant is unlikely to cause gallstone formation21.
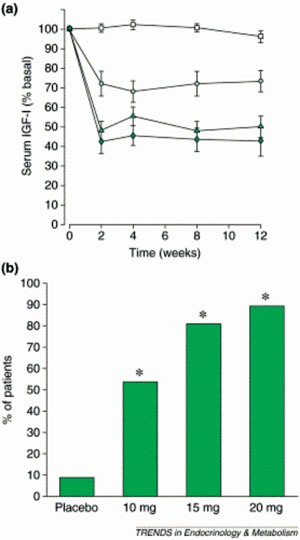
Figure 4. Effect of pegvisomant on IGF-I. (a) Serum IGF-I (expressed as % of basal value) versus time in 112 patients with acromegaly assigned to receive either placebo (squares) or one of 10 (circles), 15 (triangles) or 20 mg d-1 (diamonds) pegvisomant. (b) % of patients with a normal serum IGF-I at any stage of the same study (*, P <0.001 compared with placebo in all cases) [reprinted from (15) with permission from Elsevier].
PEGVISOMANT AND ACROMEGALY
i) Acromegaly: diagnosis and current treatment modalities
Acromegaly is the condition resulting from excessive GH and, hence, IGF-I secretion in adults, usually from a pituitary tumour. Its annual incidence is 3-4 cases per million, with a prevalence of 50-70 cases per million. It shows an equal sex distribution and the mean age at diagnosis is 42 years for men and 44 years for women22. Its clinical signs and symptoms develop insidiously, thereby causing a delay of 5-15 years in diagnosis23. These arise from the effects of excess GH/IGF-I (mainly bony overgrowth, soft tissue enlargement, hyperhydrosis, seborrhoea, sleep apnoea, kyphoscoliosis, arthropathy, carpal tunnel syndrome, proximal myopathy, cardiovascular disease with left ventricular hypertrophy, hypertension and ischaemic heart disease, peripheral insulin resistance, macroglossia, visceromegaly, colonic polyps, amenorrhea, impotence, hypercalciuria and multinodular goitre), excess prolactin (co-secretion is observed in 30% of tumours) and the tumour mass (headaches, visual field defects, cranial nerve palsy, hypopituitarism)24. Notably, younger patients tend to have more aggressive and larger tumours22. Its diagnosis is biochemically confirmed by failure of GH suppression to below 1 µg/L in response to a 75 g oral glucose load, as well as by increased serum IGF-I levels25. Acromegaly is associated with a two-to-threefold increase in mortality with major causes of death being cardiovascular, cerebrovascular and respiratory diseases26. It is of note that the relationship between acromegaly and incidence of malignancy has not been fully elucidated and, although most series suggest an increased risk of cancer, the largest cohort did not show an elevated overall cancer mortality rate; nevertheless, it did demonstrate an increased risk of colonic cancer24. It has been suggested that pre- and post-treatment levels of GH, duration of symptoms prior to diagnosis, older age at diagnosis and the presence of cardiovascular disease and hypertension are determinants of morbidity and mortality outcomes27.
The goals of therapy include a decrease or stabilization of pituitary tumour size, reversal of symptoms and signs, restoration of abnormal biochemistry to levels that reverse excess mortality and prevention of recurrence. The above targets should be achieved without adversely affecting pituitary function26. Epidemiological data suggest that when mean GH levels are lowered to <5 mU/L (2.5 µg/L), mortality is comparable to that of the general population28,29. Similar results are obtained after IGF-I normalization by transsphenoidal surgery and adjuvant therapy30. According to the 1999 consensus meeting on the treatment of acromegaly, the criteria for its biochemical remission are nadir GH levels after an oral 75 g glucose load of <1 µg/L and circulating IGF-I within the age and sex-adjusted normal range25.
Current treatment modalities include surgery, radiotherapy and medical therapy. The choice of therapy depends on the age and general health of the patient, the severity and complications of the diagnosis, as well as the dangers associated with each treatment26. Surgery, unless contraindicated, remains the primary mode of therapy providing the most rapid way of decreasing GH and IGF-I levels. Transsphenoidal adenomectomy is the preferred procedure, but the transcranial approach is occasionally necessary when there is a large suprasellar extension. The outcome of surgery depends on the pre-treatment GH levels, the size and degree of extension of the tumour, as well as the skill and experience of the neurosurgeon31-34. Although the criteria used to judge success differ widely, the best available figures in the best surgical hands suggest that 70-90% of patients with microadenomas and 45-50% of those with macroadenomas achieve safe GH levels35. It should, however, be pointed out that since 60% of the acromegalics present with macroadenomas24, a substantial number of patients will continue to have high GH and IGF-I levels after surgery. The complications of surgery include hypopituitarism, transient or permanent diabetes insipidus, cerebrovascular fluid leaks, haemorrhage and meningitis24. The recurrence rates have been reported at between 5-10%30,36. Finally, although a number of retrospective studies suggest a beneficial role for preoperative somatostatin analogues in the surgical outcome, definite data from randomized trials are not yet available37,38.
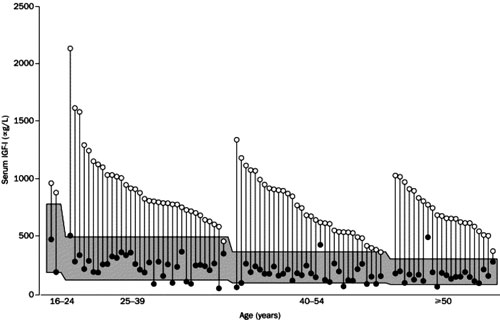
Figure 5. Baseline and lowest values of individual serum IGF-I levels in 90 patients after daily treatment with pegvisomant for 12 months or more. The shaded area represents the age-adjusted normal range for IGF-I. Ninety-seven % of the patients achieved normal IGF-I concentrations [reprinted from (53), with permission from Elsevier].
Conventional three-field pituitary radiotherapy is usually reserved for patients after unsuccessful transsphenoidal surgery and/or for treatment of disease poorly responsive to medical therapy. It prevents further tumour growth in more than 99% of cases, but it has a slow onset of action (at 10 years around 50% of patients achieve a GH level of <5 mU/L and a normal IGF-I) and it may be ineffective in normalizing IGF-I levels24,39,40,41. It may also be associated with a high occurrence of hypopituitarism (gonadal, adrenal and thyroid deficiency occurs in 50%, 35% and 35% of the cases, respectively, at 10 years)24. The major determinant of predicting response is the pre-irradiation GH level42. Rare late complications include visual deficits, second malignancy in the field of radiotherapy and possible neuropsychological changes43. Stereotactic radiotherapy (using gamma knife) delivers a more targeted radiation dose with minimal dosing to the surrounding tissues. Although longer follow-up studies are required to assess its efficacy and adverse effects, the available studies suggest that it may lead to more rapid biochemical control44,45.
Medical therapy includes somatostatin analogues, dopamine agonists and GH receptor antagonists. Somatostatin analogues (octreotide or the long-acting formulations octreotide LAR and lanreotide) are used as adjuvant therapy if safe GH levels are not achieved post-operatively, as primary treatment in the elderly and after radiotherapy until it effectively reduces GH levels. According to various reports, safe GH levels are observed in 33-78% of the patients and normal IGF-I in 39-72% of them26,46. Moreover, they may induce substantial tumour shrinkage in a significant number of patients47. The major factors predicting response to treatment are the tumour size, the somatostatin receptor density and the pre-treatment serum GH levels47-49. The primary side effects of somatostatin analogues are transient gastro-intestinal disturbances in 22% of patients and cholesterol gallstone development in up to 20% of them26. Dopamine agonists (e.g. bromocriptine, quinagolide and cabergoline) bind to the pituitary D2 receptors and suppress GH secretion in patients with acromegaly through an as yet unclear mechanism. They may be used as monotherapy or in combination with somatostatin analogues. They have a low success rate since less than 10% of patients treated with bromocriptine achieve normal IGF-I and <20% of them a GH concentration <5 µg/L50. Interestingly, better results are observed with cabergoline as 39% of patients may have IGF-I<300 µg/L51. They are beneficial in cases of co-existent secretion of prolactin leading to tumour shrinkage. Although they are relatively inexpensive, they have limited tolerability, mainly due to gastrointestinal symptoms and postural hypotension.
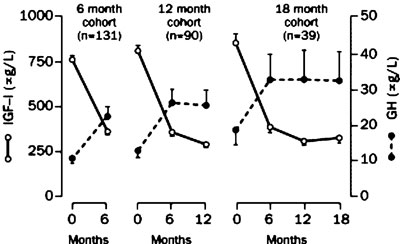
Figure 6. Serum concentrations of IGF-I and GH during treatment with pegvisomant. GH levels showed a significant increase mirroring the fall of IGF-I concentrations until the 6th month of the drug administration [reprinted from (53), with permission from Elsevier].
The above presented data support the notion that the available treatment modalities fail to achieve the currently acceptable goals of therapy in a substantial number of patients, thereby necessitating the development of new treatment options.
ii) Pegvisomant in acromegaly
Pegvisomant is the first GHR antagonist available for clinical development representing a new, promising prospect in the medical therapy of acromegaly. In contrast to the other medical treatments, its efficacy does not depend on the characteristics of the tumour (expression of dopamine or fully functional somatostatin receptors). Since it inhibits GH binding to its receptor, serum GH levels cannot be used as a biochemical marker of treatment success. Instead, the goal of treatment is normalization of IGF-I levels.
Two main studies have so far been published, investigating the efficacy of pegvisomant in acromegaly. In the first 12-week, randomized, double-blind study, 112 patients with active acromegaly were assigned to receive either pegvisomant (at a daily dose of 10 mg, 15 mg, 20mg subcutaneously) or a placebo52. Ninety-three of the patients had undergone pituitary surgery (of whom 57 had also been treated with conventional radiotherapy), 6 had been irradiated without surgery, 5 had received only drug therapy and 4 had received no therapy at all. In contrast to the placebo group in which IGF-I concentrations did not change appreciably, a dose-dependent increase in the frequency of normal IGF-I concentrations was observed in the three pegvisomant groups (54%, 81% and 89%, respectively) (Figure 4). Serum levels of free IGF-I, IGFBP-3 and the acid-labile subunit of IGFBP-3 were also reduced in the pegvisomant-treated patients in a dose-dependent manner. All groups receiving pegvisomant showed amelioration of some clinical manifestations of acromegaly (soft tissue swelling, ring size, fatigue, excessive perspiration). Serum GH concentrations increased and then showed a plateau in the pegvisomant groups in a dose-dependent fashion that coincided with the magnitude and timing of the reduction of serum IGF-I levels. Interestingly, there was no significant change in the pituitary tumour volume during the study. The drug was well tolerated; apart from injection-site reactions, the only serious adverse effect observed in one patient was an increase in liver enzyme levels (>10-times the upper limit of normal) without changes in serum bilirubin or alkaline phosphatase, which returned to normal within 8 weeks after the discontinuation of pegvisomant. Rechallenge with it was associated with a further rise in liver enzymes.
A subsequent multicentre study, which was an extension of the previously described experience, provided further evidence of the efficacy of pegvisomant. It included 152 acromegalic patients, with serum IGF-I concentrations at least 1.3 times the upper limit of the age-adjusted normal range, treated for up to 18 months with a maximum dose of 40 mg/day53. Eighty-seven of 90 patients (97%) treated for 12 months or more achieved normal serum IGF-I levels (Figure 5); this was observed even in the most severe cases of acromegaly, irrespective of the pre-treatment values of GH or IGF-I. It is of note that in 11 of these 90 patients serum IGF-I concentrations fell below the lower limit of the age-adjusted normal range, with 9 of them requiring a downward titration of their dose. GH concentrations rose substantially with the time course of the increases mirroring the falls in IGF-I levels; however, after the 6th month of treatment this did not continue (Figure 6). Paired sets of baseline and follow-up scans (mean time between them 11.46 months) performed in 131 patients with adenomas >1 cm3 (notably, 78 of them were previously treated with radiotherapy) did not show a significant increase in mean tumour volume. Furthermore, changes in tumour volume were not related to the duration of therapy. It should be pointed out that two patients who were not pre-treated with radiotherapy and had large globular adenomas with impingement on the optic chiasm demonstrated progressive growth of their tumours necessitating further treatment. It is unclear whether this was part of the natural history of the growth of the tumour or growth secondary to pegvisomant administration. Interestingly, in one of them co-treatment with octreotide halted further tumour growth and resulted in an adequate control of his disease activity54. This may support the hypothesis that co-administration of pegvisomant with octreotide could result in a synergistic decrease in serum IGF-I concentrations not achieved with octreotide or pegvisomant administration alone. Furthermore, pegvisomant concentrations up to 500-fold greater than endogenous GH levels were required to control disease, a finding consistent with the notion that pegylation reduces the drug's binding potency. The most commonly reported adverse effects were infection (usually of upper respiratory tract that rarely required treatment), headache and pain. Two patients developed a >10-times the upper limit of normal increase in serum liver enzyme concentrations within 12 weeks of beginning therapy. They remained asymptomatic, except for mild fatigue, and liver enzymes returned to normal within several months after stopping the drug. The mechanism of this effect remained unclear. Finally, tachyphylaxis was not observed and low titres of antibodies to pegvisomant not compromising its efficacy were detected in 16.9% of the patients.
The efficacy of pegvisomant in achieving normal IGF-I levels has additionally been demonstrated in acromegalics resistant to standard medical therapy with dopamine agonists and/or somatostatin analogues55,56. Pegvisomant treatment in acromegaly has also been associated with improvement of various metabolic parameters; it decreases fasting serum insulin53,57,58 and glucose concentrations53,57,58, insulin resistance (calculated using HOMA) and HBA1c58. Although the mechanisms implicated in the metabolic changes consequent upon GHR blockage remain to be elucidated, the above data suggest that activation of GH receptor leads to alterations in insulin sensitivity. Improvement in insulin sensitivity and pancreatic beta cell function has been observed in acromegalics converted from depot octreotide to pegvisomant59. Moreover, pegvisomant induces further improvement in glucose homeostasis in acromegalics who already have normal IGF-I levels during previous treatment with octreotide, a finding consistent with the hypothesis that pegvisomant and octreotide affect glucose metabolism differently60. Normalization of serum IGF-I by pegvisomant in patients with acromegaly induces alterations in plasma lipids; it increases the lowered baseline serum total cholesterol and LDL concentrations, restoring the distribution of their values to that of the general population; and it causes a favourable fall in Lp(a) and raises the apo A1 levels61. It has been proposed that similar changes might not be observed with other therapies for acromegaly. Patients with acromegaly have reduced serum leptin compared with BMI-mached controls. Pegvisomant causes a significant increase in serum leptin independently of changes in BMI57.
Furthermore, it increases C-reactive protein levels, an important cardiovascular risk marker, but the pathophysiological significance of this effect is unknown62. A recent study has also shown that it induces a reduction of left ventricular hypertrophy and improves diastolic filling63.
Treatment of acromegalics with pegvisomant for a period of 12 weeks is associated with rapid reductions of bone formation and resorption markers64. Nevertheless, long-term studies are necessary to determine whether these reductions persist, as well as the net impact of decreased bone turnover upon bone mineral density.
Finally, blockage of GH action with pegvisomant in patients with active acromegaly corrects cortisol metabolism via reversal of the inhibition of 11â-hydroxysteroid dehydrogenase65.
CONCLUSIONS
Despite the existing multimodality approach, many patents with acromegaly do not achieve the goals of therapy. Pegvisomant, a new genetically engineered analogue of human GH, represents a novel, promising concept in the medical treatment of acromegaly. The small number of studies published so far suggest that it is the most potent medical therapy with greater specificity and without being dependent on the tumour characteristics. Nonetheless, its high cost is a limiting factor and further long-term data are required to confirm its high success rate as well as to answer crucial questions regarding its safety: Is the increase of GH levels accompanied by growth of these generally considered slow growing tumours? Should the reported two cases of increase in tumour volume raise significant concerns? Is continued radiological surveillance, especially for patients who have not received radiotherapy, mandatory? What is the mechanism and the relevance of the observed altered liver function tests? What is the mechanism of GH levels increase? Furthermore, although determination of serum IGF-I concentrations may be a more cost-effective test, its appropriateness for monitoring treatment success should be carefully investigated. Indeed, the limited number of studies on "safe" IGF-I values associated with restoration of mortality to normal, the IGF-I assay variability, the discrepancy between serum GH and IGF-I levels, the sex differences in IGF-I generation by a given GH stimulus and the fact that serum IGF-I is mainly a marker of hepatic IGF-I production and not a true measure of IGF-I activity raise considerable dilemmas as to using serum IGF-I as a marker of the disease activity15,66. Notably, the possibility of overtreatment should also be considered as up to 30% of patients with severe GH deficiency have a normal serum IGF-I, and its diagnosis in the pegvisomant treated patients may be difficult due to the high circulating levels of GH not allowing the use of provocative tests67.
Moreover, careful selection of the GH assays is mandatory since the high homology between pegvisomant and the wild-type GH may be responsible for detection of the drug by most routine assays producing false elevated GH levels17,19.
Finally, further studies are required to determine the value of combined treatment with pegvisomant and somatostatin analogues, particularly in terms of preventing tumour growth, to elucidate the mechanism of the metabolic alterations associated with pegvisomant therapy and to develop new algorithms for achievement of targets of therapy.
Currently, pegvisomant, which will probably receive a license for use in Europe by the end of this year, is a second line treatment for acromegaly with an adjuvant role and possibly of greater value in cases of resistance to other treatment modalities.
Niki Karavitaki is a recipient of a grant from the Hellenic Endocrine Society.
REFERENCES
1. Abdel-Meguid SS, Shieh HS, Smith WW, Dayringer HE, Violand BN, Bentle LA, 1987 Three dimensional structure of a genetically engineered variant of porcine growth hormone. Proc Natl Acad Sci USA 84: 6434-6437.
2. De Vos AM, Ultsch M, Kossiakoff AA, 1992 Human growth hormone and extracellular domain of its receptor: Crystal structure of the complex. Science 255: 306-312.
3. Kopchick JJ, Andry JM, 2000 Growth hormone (GH), GH receptor, and signal transduction. Mol Genet Metab 71: 293-314.
4. Kelly PA, Djiane J, Postel-Vinay MC, Edery M, 1991 The prolactin/growth hormone receptor family. Endocr Rev 12: 235-251.
5. Cunningham BC, Ultsch M, De Vos AM, Mulkerrin MG, Clauser KR, Wells JA, 1991 Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule. Science 254: 821-825.
6. Gent J, Van Den Eijnden M, Van Kerkhof P, Strous GJ, 2003 Dimerization and signal transduction of the growth hormone receptor. Mol Endocrinol 17: 967-975.
7. Green H, Morikawa M, Nixon T, 1985 A dual effector theory of growth-hormone action. Differentiation 29: 195-198.
8. Casanueva FF, 1992 Physiology of growth hormone secretion and action. Endocrinol Metab Clin North Am 21: 483-513.
9. Chen WY, Wight DC, Wagner TE, Kopchick JJ, 1990 Expression of a mutated bovine growth hormone suppresses growth of trangenic mice. Proc Natl Acad Sci USA 87: 5061-5065.
10. Chen WY, Wight DC, Wagner TE, Kopchick JJ, 1991 Functional antagonism between endogenous mouse growth hormone (GH) and a GH analog results in dwarf trangenic mice. Endocrinology 129: 1402-1408.
11. Chen WY, Wight DC, Mehta BV, Wagner TE, Kopchick JJ, 1991 Glycine 119 of bovine growth hormone is critical for growth-promoting activity. Mol Endocrinol 5: 1845-1852.
12. Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV, Wells JA, 1992 Rational design of potent antagonists to the human growth hormone receptor. Science 256: 1677-1680.
13. Cunningham BC, Wells JA, 1992 Rational design of receptor-specific variants of human growth hormone. Proc Natl Acad Sci USA 88: 3407-3411.
14. Harding PA, Wang X, Okada S, Chen WY, Wan W, Kopchick JJ, 1996 Growth hormone (GH) and a GH antagonist promote GH receptor dimerization and internalization. J Biol Chem 271: 6708-6712.
15. Drake WM, Parkinson C, Besser GM, Trainer PJ, 2001 Clinical use of a growth hormone receptor antagonist in the treatment of acromegaly. Trends Endocrinol Metab 12: 408-413.
16. Clark R, Olson K, Fuh G, et al, 1996 Long-acting growth-hormones produced by conjugation with polyethylene glycol. J Biol Chem 271: 21969-21977.
17. Ross RJM, Leung KC, Maamra M, et al, 2001 Binding and functional studies with the growth hormone receptor antagonist, B2036-PEG (Pegvisomant), reveal effects of pegylation and evidence that it binds to a receptor dimer. J Clin Endocrinol Metab 86: 1716-1723.
18. Goffin V, Bernichtein S, Carriere O, Bennet W, Kopchick JJ, Kelly PA, 1999 The human growth hormone antagonist B2036 does not interact with the prolactin receptor. Endocrinology 140: 3853-3856.
19. Thorner MO, Strasburger CJ, Wu Z, et al, 1999 Growth hormone (GH) receptor blockage with PEG-modified GH (B2036-PEG) lowers insulin-like growth factor-I but does not acutely stimulate serum GH. J Clin Endocrinol Metab 84: 2098-2103.
20. Veldhuis JD, Bidlingmaier M, Anderson SM, Wu Z, Strasburger CJ, 2001 Lowering total plasma insulin-like growth factor I concentrations by way of a novel, potent and se
lective growth hormone (GH) receptor antagonist, pegvisomant (B2036-Peg), augments the amplitude of GH secretory bursts and elevates basal/nonpulsatile GH release in healthy women and men. J Clin Endocrinol Metab 86: 3304-3310.
21. Parkinson C, Drake WM, Roberts ME, Meeran K, Besser GM, Trainer PJ, 2002 A comparison of the effects of pegvisomant and octreotide on glucose, insulin, gastrin, cholecystokinin, and pancreatic polypeptide responses to oral glucose and a standard mixed meal. J Clin Endocrinol Metab 87: 1797-1804.
22. Duncan E, Wass JAH, 1999 Investigation protocol: acromegaly and its investigation. Clin Endocrinol 50: 285-293.
23. Molitch ME, 1992 Clinical manifestations of acromegaly. Endocrinol Metab Clin North Am 21: 597-615.
24. Wass JAH, Turner HE, 2002 Acromegaly In: Wass JAH, Shalet SM (eds) Oxford Textbook of Endocrinology and Diabetes, Oxford University Press, Oxford; pp, 181-192.
25. Guistina A, Barkan A, Casanueva FF, et al, 2000 Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab 85: 526-529.
26. Sheppard MC, 2003 Primary medical therapy for acromegaly. Clin Endocrinol 58: 387-399.
27. Rajasoorya C, Holdaway IM, Wrighton P, Scott DJ, Ibbertson HK, 1994, Determinants of clinical outcome and survival in acromegaly. Clin Endocrinol 41:95-102.
28. Bates AS, Van' t Hoff W, Jones JM, Clayton RN, 1993 An audit of outcome of treatment in acromegaly. Q J Med 86:293-299.
29. Orme SN, Menaly RJQ, Cartwright RA, Belchetz PE, 1998 Mortality and cancer incidence in acromegaly. J Clin Endocrinol Metab 83: 3419-3426.
30. Swearingen B, Barker FG, Katznelson L, et al, 1998, Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab 83: 3419-3426.
31. Sheaves R, Kaltsas GA, Isidori AM, et al, 2001 Predictors of the outcome of surgical treatment in acromegaly and the value of the mean growth hormone day curve in assessing postoperative disease activity. J Clin Endocrinol Metab 86: 1645-1651.
32. Jenkins P, Blackburn P, Huneidi AH, et al, 1996 Outcome of transsphenoidal surgery for acromegaly using strict criteria for surgical cure. Clin Endocrinol 45: 407-413.
33. Ahmed S, Elsheikh M, Stratton Im, Page RC, Adams CB, Wass JAH, 1999 Outcome of transsphenoidal surgery for acromegaly and its relationship to surgical experience. Clin Endocrinol 50: 561-567.
34. Gittoes NJ, Sheppard MC, Johnson AP, Stewart PM, 1999 Outcome of surgery for acromegaly—the experience of a dedicated pituitary surgeon. QJM 92: 741-745.
35. Wass JAH, Turner HE, Adams CBT, 1999 The importance of locating a good pituitary surgeon. Pituitary 2: 51-54.
36. Freda PU, Wardlaw SL, Post KD, 1998 Long-term endocrinological follow-up evaluation in 15 patients who underwent transsphenoidal surgery for acromegaly. J Neurosurg 89: 353-358.
37. Colao A, Ferone D, Cappabianca P, et al, 1997. Effect of octreotide pretreatment on surgical outcome in acromegaly. J Clin Endocrinol Metab 82: 3308-3314.
38. Abe T, Ludecke DK, 2001 Effects of preoperative octreotide treatment on different subtypes of 90 GH-secreting pituitary adenomas and outcome in one surgical centre. Eur J Endocrinol 145: 137-145.
39. Barkan AL, Halasz I, Dornfeld KJ et al, 1997 Pituitary irradiation is ineffective in normalizing plasma insulin-like growth factor I in patients with acromegaly. J Clin Endocrinol Metab 82: 3187-3191.
40. Van der Lely AJ, de Herder WW, Lamberts SW, 1997 The role of radiotherapy in acromegaly. J Clin Endocrinol Metab 82: 3185-3186.
41. Powell JS, Wardlaw SL, Post KD, Freda PU, 2000 Outcome of radiotherapy for acromegaly using normalization of insulin-like growth factor I to define cure. J Clin Endocrinol Metab 85: 2065-2071.
42. Barrande G, Pittino-Lungo M, Cote J, et al, 2000 Hormonal and metabolic effects of radiotherapy in acromegaly: long-term results in 128 patients followed in a single centre. J Clin Endocrinol Metab 85: 3779-3785.
43. Wass JAH, 2003 Radiotherapy in acromegaly: a protagonist's viewpoint. Clin Endocrinol 58: 128-131.
44. Zang N, Pan L, Wang EM, et al, 2000 Radiosurgery for growth hormone-producing pituitary adenomas. J Neurosurg 93: Suppl 3: 6-9.
45. Landolt AM, Haller D, Lomax N, et al, 1998 Stereotactic radiosurgery for recurrent surgical treated acromegaly: comparison with fractionated radiotherapy. J Neurosurg 88: 1002-1008.
46. Guisti M, Gassoni G, Cuttica CM, Girdano G, 1996 Effectiveness and tolerability of slow release lanreotide treatment in active acromegaly: six-month report on an Italian Multicenter Slow Release Lanreotide Study Group. J Clin Endocrinol Metab 81: 2089-2097.
47. Bevan JS, Atkin SL, Atkinson AB, et al, 2002. Primary medical therapy for acromegaly: an open prospective multicentre study of the effects of subcutaneous and intramuscular slow-release octreotide on growth hormone, insulin-like growth factor-I and tumour size. J Clin Endocrinol Metab 87: 4554-4563.
48. Newman CB, Melmed S, Snyder PJ, et al, 1995 Safety and efficacy of long-term octreotide therapy of acromegaly: results of a multicenter trial in 103 patients - a clinical research center study. J Clin Endocrinol Metab 80: 2768-2775.
49. Reubi JC, Landolt AM, 1989 The growth hormone responses to octreotide in acromegaly correlate with adenoma somatostatin receptor status. J Clin Endocrinol Metab 68: 844-950.
50. Melmed S, Jackson I, Kleinberg D, Klibanski A, 1998 Current treatment guidelines for acromegaly. J Clin Endocrinol Metab 83: 2646-2652.
51. Abs R, Verhelst J, Maiter D, et al, 1998 Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab 83: 374-378.
52. Trainer PJ, Drake WM, Katznelson L, et al, 2000 Treatment of acromegaly with the growth hormone-receptor
antagonist pegvisomant. N Engl J Med 342: 1171-1177.
53. Van der Lely AJ, Hutson R, Trainer PJ, et al, 2001 Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 358: 1754-1759.
54. Van der Lely AJ, Muller AF, Janssen JA, et al, 2001 Control of tumour size and disease activity during co-treatment with octreotide and the growth hormone receptor antagonist pegvisomant in an acromegalic patient. J Clin Endocrinol Metab 86: 478-481.
55. Herman-Bonert VS, Zib K, Scarlett JA, Melmed S, 2000 Growth hormone receptor antagonist therapy in acromegalic patients resistant to somatostatin analogs. J Clin Endocrinol Metab 85: 2958-2961.
56. Drake WM, Parkinson C, Akker SA, et al, 2001 Successful treatment of resistant acromegaly with growth hormone receptor antagonist. Eur J Endocrinol 145: 451-456.
57. Parkinson C, Whatmore AJ, Yates AP, et al, 2003 The effect of pegvisomant-induced serum IGF-I normalization on serum leptin levels in patients with acromegaly. Clin Endocrinol 59: 168-174.
58. Rose RD, Clemmons DR, 2002 Growth hormone receptor antagonist improves insulin resistance in acromegaly. Growth Horm IGF Res 12: 418-424.
59. Drake WM, Rowles SV, Roberts ME, et al, 2003 Improvement in insulin sensitivity in patients with acromegaly converted from Depot Octreotide (Sandostatin LAR) to pegvisomant. Endocrine Society's 85th Annual Meeting, Philadelphia P1-633.
60. Clemmons DR, Barkan AL, Melmed S, et al, 2003 Pegvisomant improves glucose homeostasis in patients with acromegaly previously treated with octreotide LAR. Endocrine Society's 85th Annual Meeting, Philadelphia P2-341.
61. Parkinson C, Drake WM, Wieringa G, Yates AP, Besser GM, Trainer PJ, 2002 Serum lipoprotein changes following IGF-I normalization using a growth hormone receptor antagonist in acromegaly. Clin Endocrinol 56: 303-311.
62. Sesmilo G, Fairfield WP, Katznelson L, et al, 2002 Cardiovascular risk factors in acromegaly before and after normalization of serum IGF-I levels with the GH antagonists pegvisomant. J Clin Endocrinol Metab 87: 1692-1699.
63. Collao A, Pivonello R, Gaderisi M, et al, 2003 Short-term effects of pegvisomant on the acromegalic cardiomyopathy: a 6-month prospective study by echocardiography. Endocrine Society's 85th Annual Meeting, Philadelphia P1-626
64. Fairfield WP, Sesmilo G, Katznelson L, et al, 2002 Effects of growth hormone receptor antagonist on bone markers in acromegaly. Clin Endocrinol 57: 385-390.
65. Trainer PJ, Drake WM, Perry LA, Taylor NF, Besser GM, Monson JP, 2001 Modulation of cortisol metabolism by the growth hormone receptor antagonist pegvisomant in patients with acromegaly. J Clin Endocrinol Metab 86: 2989-2992.
66. Brabant G, 2003 Insulin-like growth factor-I: marker for diagnosis of acromegaly and monitoring the efficacy of treatment. Eur J Endocrinol 148: 15-20.
67. Trainer PJ, 2002 A giant' s step forward. Clin Endocrinol 56: 423-425.
Address correspondence and requests for reprints to:
John AH Wass, Professor of Endocrinology, Department of Endocrinology, the Oxford Centre for Diabetes,
Endocrinology and Metabolism, Churchill Hospital, Old Road, Headington, Oxford, OX3 7LJ, UK,
Tel.: +44 1865857310, Fax: +44 1865857368, e-mail: john.wass@noc.anglox.nhs.uk
Received 03-10-03, Revised 10-11-03, Accepted 05-12-03