1Department of Endocrinology & Diabetes, 251 Hellenic Air Force & VA General Hospital, Athens, Greece, 2Department of Endocrinology & Metabolic Diseases, Leiden University Medical Center, Leiden, The Netherlands
Hypercalcaemia results from the failure of renal calcium excretion to compensate increased influx of calcium into the circulation from the intestine, the kidneys and the skeleton. Hypercalcaemia is a common metabolic abnormality of varying severity that can be adequately diagnosed and treated. Primary hyperparathyroidism and malignant neoplasms are responsible for >90% of all cases. The management of hypercalcaemia depends on the underlying cause and involves approaches aiming at reducing serum calcium concentrations and correcting associated metabolic disturbances. A number of pharmacological interventions are currently available for the treatment of hypercalcaemia related to malignancy as well as other forms of hypercalcaemia. Knowledge of the pathophysiology of hypercalcaemia and of the properties of these interventions is essential for the successful management of affected individuals. We herein review available therapeutic interventions for hypercalcaemia of varying aetiology.
Bisphosphonates, Calcimimetics, Hypercalcaemia, Hyperparathyroidism, Malignancy, Parathyroid hormone, Parathyroid hormone related protein
INTRODUCTION
Calcium is a critical ion for many physiological processes. Circulating calcium concentrations fluctuate within a narrow range due to the tight regulation of the fluxes at the three main sites of calcium transport (intestine, bone, kidney) mainly by the calciotropic hormones.1-3 Serum total calcium concentrations, adjusted to serum albumin values, range between 8.5 and 10.5 mg/dl (2.1-2.5 mmol/l). The adjustment to serum albumin is accomplished by adding or subtracting 0.08 mg/dl (0.02 mmol/l) to the measured total serum calcium for every 1.0 gr/l of albumin below or above 40 gr/l albumin, respectively. Measurement of ionized serum calcium concentration is rarely needed in clinical practice. Hypercalcaemia develops when the amount of calcium entering the circulation from the sites of its transport exceeds the capacity of the kidney to excrete it.
Symptoms of hypercalcaemia are non-specific or can be related to the underlying disease (Table 1 ). Furthermore, the severity of the symptoms depends on the level of serum calcium and on the rate of its increase. The most frequent causes of hypercalcaemia are primary hyperparathyroidism and malignant diseases in out-patients and in-patients, respectively.4 Other, less frequent causes of hypercalcaemia are shown in Table 2 .
In this article we briefly discuss the pathophysiology of hypercalcaemia and review current approaches to its management.
MALIGNANCY-ASSOCIATED HYPERCALCAEMIA (MAH)
Hypercalcaemia occurs in about 20-30% of patients with malignancies during the course of the disease, is associated with poor prognosis and can be a medical emergency.4-6 In malignant diseases hypercalcaemia is mainly due to increased bone resorption by factors released by the tumour cells locally or systemically (Figure 1). Various pathophysiological classifications of malignancy-associated hypercalcaemia have been proposed. One such classification recognizes four separate types:4,7
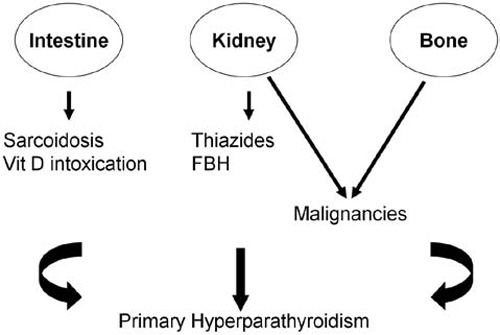
Figure 1. Schematic representation of the pathophysiology of hypercalcaemia in different conditions, depicting the contribution of intestine, kidney and bone to its development. FBH: Familial Benign Hypercalcaemia.
1. Local Osteolytic Hypercalcaemia (LOH). This is encountered in patients with extensive skeletal involvement, such as multiple myeloma and breast cancer, and is the most frequent cause of LOH among solid tumours.7 The primary mechanism of hypercalcaemia in LOH is increased bone resorption. Once in the bone microenvironment, tumour cells release factors which stimulate the formation and/or the activity of osteoclasts. Bone matrix components, which are liberated during resorption, act chemotactically for tumour cells or alter their characteristics and increase their metastatic potential thus amplifying the metastatic process. For example, Parathyroid Hormone related Peptide (PTHrP), which is expressed by the majority of bone metastases from breast cancer, stimulates the production of the receptor activator of nuclear factor-κΒ ligand (RANKL) by osteoblastic/stromal cells in the bone/bone-marrow microenvironment, thereby enhancing osteoclastogenesis and bone resorption. This, in turn, leads to release of Transforming Growth Factor β (TGF-β) from bone which acts on PTHrP receptors, present on the cancer cells to produce more PTHrP. In addition, factors that suppress osteoblast function (Dkk1) may also be secreted and contribute to increased bone resorption, as shown in multiple myeloma.8 Increased bone resorption, biochemically assessed, has also been shown to predict the development of skeletal complications in cancer patients.
2. Humoral Hypercalcaemia of Malignancy (HHM). This type results mainly from the systemic secretion of PTHrP by squamous carcinomas (esophagus, lung, cervix, vulva, head and neck, etc) as well as renal, bladder, ovarian or even breast carcinomas and pancreatic neuroendocrine tumours.7,9,10 PTHrP induces hypercalcaemia by increasing osteoclastic bone resorption and the renal tubular reabsorption of calcium. Biochemical findings resemble those of primary hyperparathyroidism (PHP) but serum PTH is markedly suppressed.11 Other as yet unidentified humoral factors may also induce hypercalcaemia in some patients with malignant diseases.
3. Vitamin D-mediated hypercalcaemia. This type is due to ectopic production of 1,25-dihydroxyvitamin D [1,25(OH)2D] by some lymphomas and ovarian dysgerminomas.4,7,12-14 It is a rare cause of MAH and is considered the malignant counterpart of sarcoidosis.4,7,12,13
4. Ectopic secretion of authentic PTH by the tumour. It is an extremely rare cause of hypercalcaemia in patients with cancer.7,15,16 About ten cases of ectopic hyperparathyroidism have been reported,7,16 but the condition should be considered in the differential diagnosis of hypercalcaemia in patients with cancer and elevated serum PTH.
TREATMENT OF MAH
Development of hypercalcaemia in patients with malignant diseases is a poor prognostic sign and can be fatal if left untreated. The management of MAH aims at reducing serum calcium concentrations and correcting the associated metabolic disturbances but it is not curative. It consists of general and specific measures.
General measures
The first step in the management of MAH is volume repletion with physiological saline (NaCl 0.9%). Hypercalcaemia has a potent diuretic effect leading to volume depletion and dehydration. To protect against sodium loss, the kidney increases its tubular reabsorption. However, this is associated with increased tubular reabsorption of calcium which further worsens the hypercalcaemia. Administration of physiological saline, which interrupts this sequence of events, is the cornerstone in the treatment of hypercalcaemia of any cause.1,4 Saline can be continuously infused (3-6 liters over 24-48 hours) and can lower serum calcium concentration by approximately 1.0-3.0 mg/dl (0.25mmol/L-0.75mmol/l) by decreasing the concomitant reabsorption of sodium and calcium in both the proximal and distal renal tubule and enhancing urinary calcium excretion by increasing its glomelurar filtration rate. However, volume repletion is usually not enough to normalize serum calcium concentration and should be followed by more specific interventions. Care should be taken in the administration of physiological saline in elderly patients with compromised cardiac and/or renal function because there is the possibility of volume overload.
Diuretics
Loop diuretics, particularly furosemide, are traditionally recommended in the management of MAH because they increase urinary calcium excretion. However, although in most textbooks loop diuretics are still considered the first step of treatment of MAH, scientific evidence to support their use is lacking.17 In addition, if loop diuretics are given to patients who are not volume replete, they may even worsen the hypercalcaemia because of their concomitant natriuretic effect. The use of loop diuretics should therefore be restricted to patients at risk of volume overload.
Bisphosphonates
Bisphosphonates are currently the treatment of choice for MAH due to their inhibitory action on bone resorption.18,19 MAH was one of the first clinical applications of bisphosphonates and currently two bisphosphonates (zoledronate, pamidronate) are approved for this indication in the USA, while in Europe clodronate and ibandronate are also approved.1,20-23 Bisphosphonates are given by single intravenous infusion over a period of between 15 minutes and 4 hours depending on the bisphosphonate and in the majority of patients normal serum calcium concentrations are obtained within seven days following the bisphosphonate administration. The recommended doses are: clodronate 300mg, pamidronate 90 mg, ibandronate 6 mg and zoledronate 4 mg, as a single infusion. There are only a few head-to-head studies of bisphosphonates in the treatment of MAH. In an analysis of two randomized, controlled clinical trials, zoledronate was reported to be more effective than pamidronate in reducing serum calcium concentrations in patients with MAH.24 In addition, its effect on serum calcium concentrations appeared to last longer. The clinical significance of the relatively small differences between the two bisphosphonates has, however, been questioned.1,4 It should be noted that the effect of bisphosphonates on serum calcium concentrations is apparent after 24-48 hours.25 In life-threatening hypercalcaemia, when a rapid decrease of serum calcium concentrations is needed, high doses of parenteral calcitonin can be given concomitantly with the bisphosphonate every 6 to 8 hours during the first 24 hours. With the exception of such cases, calcitonin has currently no place in the management of MAH. The beneficial effect of bisphosphonates on serum calcium concentrations can be sustained from several days to several weeks.1 In the case of an incomplete response, the infusion can be repeated after at least one week.
Adverse effects of aminobisphosphonates include flu-like symptoms due to the induction of an acute phase reaction during the first administration of these agents. This reaction is transient and symptoms can be ameliorated with acetaminophen or paracetamol. Mild, short-term hypophosphataemia and asymptomatic hypocalcaemia may also occur. Bisphosphonates should be used with care in patients with renal insufficiency and recommendations on the dose and rate of infusion of every bisphosphonate should be strictly followed. Rapid infusion of the older bisphosphonates may cause renal damage, while doses of newer bisphosphonates higher than those recommended may adversely affect renal function.4 Osteonecrosis of the jaw (ONJ) which has been reported in patients with malignant diseases and bone metastases treated with intravenous bisphosphonates, is related to the total dose and the duration of treatment with bisphosphonates. It is, therefore, highly unlikely to occur after treatment of patients with MAH.
In HHM, the effect of bisphosphonates is usually short-lived and may also be incomplete due to the contribution of coexisting skeletal and renal components to the hypercalcaemia. The renal component includes the increased distal tubular calcium reabsorption driven by PTHrP, which does not respond to bisphosphonates.7
Steroids
Glucocorticoids have been used in the past to treat hypercalcaemia, especially in patients with hematologic malignancies. Today their use is restricted to the treatment of hypercalcaemia due to ectopic production of 1,25(OH)2D.26 Glucocorticoids are rarely efficacious in hypercalcaemia of solid tumours.1 Prednisone (30 mg daily) or hydrocortisone (120 mg daily per os, or parenterally) are commonly used in the treatment of hypercalcaemia resulting from endogenous production of 1,25(OH)2D. The circulating 1,25(OH)2D concentrations decrease usually within three days followed by a decrease in serum calcium concentrations.26 Nevertheless, steroid therapy may not always be appropriate for these diseases and alternative treatments have been suggested, including chloroquine or hydroxychloroquine and ketoconazole.26-28 There is limited experience in the use of these agents as calcium-lowering therapies and they should be given only in cases of severe hypercalcaemia that does not respond to steroids or when steroids are contraindicated.
Experimental treatments
Experimental therapies for MAH include agents that interfere with the RANKL signalling pathway such as monoclonal antibodies to RANKL or osteoprotegerin (a decoy receptor of RANKL).29,30 The rationale for their use is based on their inhibitory effect on bone resorption. This effect is exerted through prevention of recruitment and of differentiation of osteoclasts and a decrease of their life-span. Monoclonal antibodies against PTHrP have also been used.31,32 It is still unknown whether these agents will prove safe and effective in humans with MAH and if they will reverse hypercalcaemia more efficiently and at less expense than the potent bisphosphonates.
Hemodialysis
Kidney replacement therapy against a dialysate containing little or no calcium is effective in patients with severe MAH and renal failure, thus precluding aggressive volume repletion and, probably, bisphosphonates.4 However, there are no specific guidelines for such an approach.
Anti-tumour therapy
As already mentioned, the effect of anti-hypercalcaemic therapy of patients with MAH is temporary.6 Moreover, in most of the cases hypercalcaemia is a sign of progression of the tumour and specific anti-tumour therapy should be promptly initiated. Control of calcium levels can provide the time needed for the anti-tumour treatment to work. Finally, optimal treatment of MAH should always be tailored to the needs of the individual patient and initiation of aggressive anti-hypercalcaemic therapy in “terminal” cancer cases should be carefully considered.4
BENIGN HYPERCALCAEMIAS
Primary Hyperparathyroidism (HPT)
Classically, HPT is distinguished into primary, secondary and tertiary. In clinical practice, the finding of raised serum calcium concentrations with increased or non-suppressed serum PTH values is indicative of autonomous HPT (primary, tertiary), whereas secondary HPT is defined by increased PTH in the presence of normal (or low) serum calcium concentrations. Primary HPT is the most frequent cause of benign hypercalcaemia, also being one of the most common of the endocrinopathies with an incidence ranging from 1 in 500 to 1 in 1000 subjects. It can occur at all ages, but it is most frequent during the sixth decade of life with a female to male ratio of 3:1. It is rather uncommon in younger ages and, if present, the possibility of its being part of multiple endocrine neoplasia (MEN) I or II should be considered. In about 80% of the cases, HPT is due to a benign single adenoma; four-gland hyperplasia occur in approximately 15-20% of the cases, either sporadically or within the context of MEN; parathyroid carcinomas are rare, accounting for <0.5% of all HPT cases. PTH increases serum calcium concentrations by acting directly on bone and kidney (increased bone resorption and renal tubular reabsorption of calcium, respectively) and indirectly on the intestine [stimulation of the production of 1,25(OH)2D] (Figure 1).
The classical clinical manifestations of primary HPT are nephrolithiasis and bone disease. However, “osteitis fibrosa cystica”, as it was originally described, is now encountered in <5% of cases, while the incidence of kidney stones has declined from 1/3 to 1/5 of the cases. Osteoporosis remains one of the most frequently described manifestations of primary HPT and many cases are discovered during the initial investigation of low bone mineral density. Moreover, severe neuromuscular symptoms, as described earlier, are no longer prominent, although vague symptoms such as easy fatigue, a sense of weakness, or even an intellectual weariness and a sense of less sharp cognitive function are rather common.33 Whether these are due to raised serum calcium concentrations, to elevated PTH or both is not known. Gastrointestinal manifestations are no longer prevalent in HPT with the exception of patients with MEN. Asymptomatic hypercalcaemia is currently the most common presentation of HPT and the vast majority of patients are diagnosed accidentally following a routine laboratory investigation. In addition, cases of normocalcaemic HPT are now being described with increasing frequency, representing either a new clinical phenotype or simply patients with an early diagnosis.34 In such cases, vitamin D deficiency/insufficiency [serum 25(OH)D <50 nmol/l (20 ng/ml)] should always be ruled out and, if present, re-evaluation of these individuals must take place after repletion with vitamin D and/or the correction of other causes of secondary HPT.33 In the differential diagnosis of HPT, hypercalcaemic disorders in which PTH may be increased should be considered. These are familial benign hypocalciuric hypercalcaemia (FBHH) and use of lithium or thiazide diuretics. Personal and family medical history can assist the diagnosis, while PTH and calcium can be re-assessed after 2-3 months of drugs withdrawal. Differential diagnosis of primary HPT and HHM presents no difficulties as there is no cross-reactivity of PTHrP in current PTH assays.
Treatment of primary HPT
Primary HPT is a disease that can be cured by the surgical removal of the hyper-functioning parathyroid tissue. Controversies regarding the surgical approach concern the timing of the intervention rather than its efficacy. To address these controversies, a series of Consensus conferences, which started in 1990, analysed available data and developed guidelines for the management of primary HPT. In symptomatic primary HPT (overt bone and/or renal disease, episodes of life-threatening hypercalcaemia), the appropriate treatment is the surgical excision of the abnormal parathyroid tissue. The current criteria for surgery of patients with asymptomatic primary HPT are: serum calcium >1 mg/dl (0.25 mmol/l) above the upper limit of the local reference range; hypercalciuria of >400 mg/day (10 mmol/d); reduced creatinine clearance of >30% compared to sex- and age-matched controls; age <50 years; BMD T-score <-2.5 determined by DXA scan at any site (forearm, lumbar spine, hip).35 The most recent criteria, however, do not include hypercalciuria as an indication for surgery, while a creatinine clearance less than 60ml/min is now considered an indication for surgery.36
Surgical treatment of HPT requires expertise due to the variability of the locations where pathological parathyroid glands can be found and the difficulty in identifying them. Preoperative localization studies have become very popular and many patients are routinely tested prior to surgery. However, an experienced parathyroid surgeon will identify and remove the abnormal parathyroid tissue in >95% of neck surgery naive patients.33 This figure of successful localization is almost never achieved by localization studies and there is little evidence that these can shorten the operation time or prevent operation failures. Preoperative localization of abnormal parathyroid tissue should therefore be restricted to cases with previous neck surgery. Transient mild, and often asymptomatic, hypocalcaemia may occur within the first few days following successful surgery, which usually does not require specific treatment. Although rarely seen today, prolonged symptomatic hypocalcaemia resulting from rapid deposition of calcium and phosphate into bone (“hungry bone” syndrome) will need oral or parenteral calcium supplementation with active metabolites of vitamin D (alpha calcidiol, calcitriol).33
Medical treatment
General measures
A number of patients with primary HPT and hypercalcaemia are not candidates for surgery and should be treated conservatively. Routine medical follow-up includes yearly assessment of bone mineral density at all sites and measurement of serum creatinine and calcium twice yearly.35 In addition, drugs that increase serum calcium concentrations (e.g. thiazides, lithium) should be avoided and adequate hydration must be recommended. The influence of calcium intake on serum calcium concentrations in patients with HPT remains controversial. A daily calcium intake of 800-1000mg is indicated because low calcium diets may further enhance PTH secretion, while high calcium intakes may increase serum calcium concentrations, especially among patients with elevated 1,25(OH)2D. Low doses of vitamin D (400-800 u/d) should be given to patients with vitamin D deficiency/insufficiency.37
Bisphosphonates
Bisphosphonates have been used in the medical management of patients with primary HPT; etidronate was ineffective, while clodronate resulted in a temporary reduction of serum calcium concentrations.38 Alendronate has been more extensively studied and either a small effect on serum calcium39,40 or no change at all have been reported.41,42 Within a similar context, PTH was reported either to increase39,42,43 or to remain unchanged.40 A multicenter, double-blind, randomized, placebo-controlled study reported no effect of alendronate on either serum calcium or PTH levels.44 However, consistent with its mechanism of action, alendronate had a major effect in reducing biochemical markers of bone turnover and improving bone mineral density of the lumbar spine and the total hip. The increase in bone mineral density has been a consistent finding of all studies using alendronate.39-41 Similarly, risedronate reduced fasting serum calcium concentrations and biochemical markers of bone turnover in a small series of patients with primary HPT; additional studies are required to evaluate the long-term efficacy of this bisphosphonate in HPT.37,45 The use of intravenous bisphosphonates (pamidronate, zoledronate) in primary HPT is restricted to the treatment of acute hypercalcaemia,33 as it occurs, for example, in parathyroid crisis, which is associated with serum calcium concentrations greater than 15 mg/dl (3.75 mmol/l) and rapid deterioration of central nervous system, cardiac, gastrointestinal and renal function. Such cases can be fatal and neck surgery should be performed as soon as possible.46 It should be noted that there are no data on the efficacy of yearly infusions of zoledronate as used in the treatment of osteoporosis.
From the results of available studies and the pathophysiology of the disease, it can be concluded that bisphosphonates given at doses used in the treatment of osteoporosis prevent bone loss and improve bone mineral density in patients with primary HPT but have no effect on PTH secretion or serum calcium concentrations.
Estrogens, raloxifene
Similar to bisphosphonates, estrogens decrease bone resorption and improve bone mineral density in postmenopausal women with primary HPT but have no effect on PTH levels. Changes in serum calcium concentrations are minimal and a decrease of approximately 0.5 mg/dl (0.12mmol/l) has been reported.33,37 However, the risks associated with estrogen use prevent their application to the long-term treatment of postmenopausal women with primary HPT. Limited data with raloxifene have shown similar results.47 Calcimimetics The minute-to-minute regulation of PTH secretion by calcium is mediated by the parathyroid cell plasma membrane Ca-Sensing Receptor (CaSR) which detects ambient serum Ca++.48 Increased serum calcium suppresses parathyroid CaSR signalling, resulting in diminished PTH secretion, while it stimulates distal nephron CaSR to reduce renal tubular calcium reabsorption. The net effect of these hypercalcaemia-driven changes is the lowering of serum calcium concentrations. In patients with primary HPT, the sigmoidal curve, which describes the relationship between serum calcium and PTH, is shifted to the right, resulting in decreased responsiveness of the parathyroid cells to the elevated calcium concentrations. Consequently, increased PTH secretion is observed despite hypercalcaemia.49 Calcimimetics are drugs which increase the sensitivity of the CaSR in parathyroid cells to circulating calcium, leading to increased intracellular calcium and subsequently reduction of PTH secretion (Figure 2).50 Cinacalcet is an oral calcimimetic that interacts with the transmembrane domain of the CaSR and increases its affinity for calcium. Cinacalcet was shown to reduce PTH secretion and the risk of adverse clinical outcomes in patients with secondary hyperparathyroidism due to renal failure51,52 and is currently approved for this indication. 
Figure 2. Left panel: Schematic representation of the Calcium Sensing Receptor (CaSR) of parathyroid cell. Right panel: The action of calcimimetics on CaSR.
Cinacalcet has been studied in patients with primary HPT and was shown to reduce serum calcium concentrations. In a small randomized, placebo-controlled study, cinacalcet (30mg, 40mg, or 50mg) was given twice daily for 15 days and was found to normalize serum calcium concentration within one day after its administration and to decrease serum PTH 2-4 hours after dosing; these effects were sustained throughout the period of treatment.49 While there was no consistent dose-response, cinacalcet doses of 40 and 50 mg twice daily resulted in larger reductions in serum calcium concentrations.49 Based on these findings, a larger randomized, placebo-controlled study was subsequently conducted in 78 patients with primary HPT.50 Cinacalcet was titrated from 30-50mg twice daily and treatment was given for one year. Normocalcaemia was achieved in up to 73% of patients and, once a steady state had been individually established, it was sustained during the whole period of the study without evidence of fluctuations. In 90% of patients, the dose of 30mg twice daily was sufficient to normalize serum calcium. Serum PTH was reduced by 37% two hours after dosing and gradually returned towards pre-dose levels by eight hours. Urinary calcium excretion decreased slightly.53 Forty-five patients were enrolled in a two-year extension study in which all received cinacalcet. After three years, 87% of those who completed the study had normal serum calcium concentrations.54 Cinacalcet has also been tested in the treatment of primary HPT due to parathyroid carcinoma. In a multicenter, open-label, single-arm study, 29 patients with parathyroid carcinoma were given cinacalcet and doses were titrated within a 16-week period from 30 mg twice daily up to 90 mg four times daily.55 Serum calcium concentration was reduced by at least 1.0 mg/dl in 62% of patients, although PTH was not significantly reduced; patients with higher baseline calcium levels required higher doses of cinacalcet, while cumulative doses up to 360 mg daily (90 mg QID) were generally well tolerated.
Common adverse effects of cinacalcet include nausea, vomiting and headache. In general, it seems that cinacalcet is a promising medical treatment of primary HPT, although further studies focusing on changes in bone mineral density and fracture risk are needed.
Cinacalcet is currently approved for the medical treatment of parathyroid carcinoma when hypercalcaemia persists and a surgical approach is no longer indicated or feasible. They are also approved for the management of “intractable” HPT, defined as unresolved HPT, following unsuccessful parathyreoidectomy or contraindicated parathyroidectomy.
There is limited experience in the medical treatment of the HPT of MEN (1 or 2) but bisphosphonates and cinacalcet are expected to act as in sporadic primary HPT. Interestingly, in a recent small series, six months of treatment with depot, long-acting octreotide, controlled hypercalcaemia and hypercalciuria in 2/3 of patients with MEN1-related HPT, while PTH decreased in all patients.56 It was suggested that this response was due to a direct action of octreotide on parathyroid tumour cells mediated by binding to somatostatine receptors. This, however, has not been the experience of others and more studies are needed before a conclusion may be formed as to the efficacy of octreotide in such patients.
FAMILIAL BENIGN HYPOCALCIURIC HYPERCALCAEMIA (FBHH)
FBHH or Familial Benign Hypercalcaemia (FBH) is an autosomal dominantly inherited disorder with a lifelong high penetrance for hypercalcaemia and relative hypocalciuria, which accounts for almost 2% of asymptomatic hypercalcaemic cases.57 It is most frequently caused by heterozygous loss-of-function mutations in the gene encoding the CaSR.58,59 It is usually asymptomatic; however, the degree of hypercalcaemia varies from very modest to quite severe [12.5-14.5 mg/dl (3.12mmol/L-3.62mmol/l)]. Parathyroid function is inappropriately normal or even modestly elevated, thus reflecting the resistance of parathyroid cell to elevated serum calcium levels. The course of the disease is benign and patients should be reassured. FBHH should always be considered in the differential diagnosis of primary HPT in order to avoid unnecessary parathyroidectomy. Immediate (within one week) recurrence of hypercalcaemia occurs after a subtotal parathyroidectomy, since even a small remnant of parathyroid tissue is enough to maintain elevated calcium levels. Therefore, surgical excision is contraindicated except in rare cases of severe hypercalcaemia [>14.0 mg/dL (3.5mmol/l)] and relapsing pancreatitis. Additionally, FBHH is resistant to previously described calcium lowering agents. However, evidence from a few case reports suggests that cinacalcet might be effective.60,61
TREATMENT OF OTHER HYPERCALCAEMIAS
Granuloma-forming disorders
Extra-renal production of 1,25(OH)2D by macrophages can occur in granuloma-forming disorders62 (Table 2). Sarcoidosis is the most extensively studied of these diseases and hypercalcaemia and hypercalciuria have been reported in up to 10% and 50% of patients, respectively, at some time during the course of the disease.26 The successful management of sarcoidosis-driven hypercalcaemia requires the recognition of high risk patients. In addition, follow-up after a hypercalcaemic episode must include close monitoring of urinary calcium excretion in order to detect recurrence timely. Glucocorticoids are the basis of treatment of hypercalcaemia of sarcoidosis and other granuloma-forming diseases. Doses are the same as those used in the treatment of malignant lympho-proliferative disorders. However, infectious diseases should be very carefully treated due to a significant risk of disease exacerbation. Excessive sunlight exposure (UV light) must be avoided in order to prevent endogenous vitamin D production.
Thyrotoxicosis
Up to one half of thyrotoxic patients manifest mild hypercalcaemia ranging from 10.5mg/dL to 11.5 mg/dL (2.62mmol/L-2.87mmol/L).63 Excessive bone resorption and calcium release into the circulation is the pathophysiological mechanism of serum calcium increase. Therefore, in the absence of other calcium-increasing disorders, PTH and 1,25(OH)2D serum levels should be low in hyperthyroidism-driven hypercalcaemia. The hypercalcaemia of thyrotoxicosis is fully reversible after restoration of normal thyroid function.63
Pheochromocytoma
Co-existence of hypercalcaemia with pheochromocytoma is most frequently described in the context of MEN 2A. The cause is unknown but correction of hypercalcaemia has been reported after removal of pheochromocytoma, suggesting that tumour-derived factors or catecholamines may be responsible.63 In addition, increased PTHrP secretion, and subsequent hypercalcaemia, have been reported in a few patients with pheochromocytomas as well as in patients with other tumours of neuroendocrine origin.
Adrenal insufficiency
Adrenal insufficiency, and in particular addisonian crisis, has been linked with hypercalcaemia.63 The most probable cause of hypercalcaemia is hypovolaemia and hemoconcentration. Hypercalcaemia is corrected with fluid repletion and steroid replacement.
Milk-alkali syndrome
The milk-alkali syndrome results from the consumption of large amounts of milk and absorbable alkali, as previously used in the treatment of peptic ulcer, or large quantities of calcium carbonate providing a daily calcium intake usually in excess of 5gr.63 It is classically defined as the triad of systemic alkalosis, hypercalcaemia and renal insufficiency. Hypercalcaemia usually reverses with rehydration and discontinuation of treatment.
Vitamin D intoxication
Vitamin D intoxication may occur with the use of doses in excess of 50,000 IU per day and is associated with serum 25(OH)D concentrations higher than 150 ng/ml (374 nmol/l) and hypercalcaemia.64 Discontinuation of treatment is usually sufficient to correct the hypercalcaemia but in severe cases rehydration and a short course of glucocorticoids may be required. Reversal of hypercalcaemia is relatively slow due to the plasma half-life of 25(OH)D, which is approximately 25 days. In contrast, hypercalcaemia occurring during treatment with active vitamin D metabolites (alphacalcidol or calcitriol) reverses quickly after treatment arrest due to the short plasma half-life of these metabolites.65
CONCLUSIONS
The management of hypercalcaemia depends on the underlying cause. Volume expansion and inhibition of bone resorption are the most important medical interventions, independently of the aetiology of hypercalcaemia. Bisphosphonates are the mainstay of medical therapy for malignancy-associated hypercalcaemia and may provide the time necessary for anti-tumour therapy to work. In PTH-mediated hypercalcaemias, surgery is the treatment of choice when appropriate criteria are met. Bisphosphonates, as used in the treatment of osteoporosis, prevent bone loss in primary hyperparathyroidism but have no effect on serum calcium levels. Calcimimetics can normalize serum calcium concentrations in a large proportion of patients with primary HPT and are approved for the medical management of parathyroid carcinoma and “intractable” autonomous hyperparathyroidism. Novel therapeutic approaches directed at specific molecular targets are currently being evaluated.
REFERENCES
1. Shane E, Irani D 2006 Hypercalcaemia: Pathogenesis, Clinical Manifestations, Differential Diagnosis and Management. In: Favus MJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 176-180.
2. Parathyroid Hormone-Related Protein: Normal Physiology and its Role in Cancer. Halloran BP, Nissenson BA, (eds) 1992 Parathyroid Hormone-Related Protein: Normal Physiology and its Role in Cancer. USA: CRC Press, Boca Raton, FL.
3. Mundy GR 2008 Role of cytokines, parathyroid hormone and growth factors in malignancy. In: Bilezikian JP, Raisz LG, Rodan GA, (eds). Principles of Bone Biology, Academic Press, New York, NY, USA; pp, 827-836.
4. Stewart AF, 2005 Clinical practice. Hypercalcemia associated with cancer. N Engl J Med 352: 373-379.
5. Roodman GD, 2004 Mechanisms of bone metastasis. N Engl J Med 350: 1655-1664.
6. Ralston SH, Gallacher SJ, Patel U, Campbell J, Boyle IT, 1990 Cancer-associated hypercalcemia: morbidity and mortality. Clinical experience in 126 treated patients. Ann Intern Med 112: 499-504.
7. Horwitz MJ, Stewart AF 2006 Hypercalcemia associated with malignancy. In: Favus MJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 195-199.
8. Tian E, Zhan F, Walker R, et al, 2003 The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 349: 2483-2494.
9. Srirajaskanthan R, McStay M, Toumpanakis C, Meyer T, Caplin ME, 2008 Parathyroid hormone-related peptide-secreting pancreatic neuroendocrine tumours: case series and literature review. Neuroendocrinology 89: 48-55.
10. Hoekman K, Tjandra YI, Papapoulos SE, 1991 The role of 1,25-dihydroxyvitamin D in the maintenance of hypercalcemia in a patient with an ovarian carcinoma producing parathyroid hormone-related protein. Cancer 68: 642-647.
11. Schweitzer DH, Hamdy NA, Frolich M, Zwinderman AH, Papapoulos SE, 1994 Malignancy-associated hypercalcaemia: resolution of controversies over vitamin D metabolism by a pathophysiological approach to the syndrome. Clin Endocrinol (Oxf) 41: 251-256.
12. Seymour JF, Gagel RF, Hagemeister FB, Dimopoulos MA, Cabanillas F, 1994 Calcitriol production in hypercalcemic and normocalcemic patients with non-Hodgkin lymphoma. Ann Intern Med 121: 633-640.
13. Evans KN, Taylor H, Zehnder D, et al, 2004 Increased expression of 25-hydroxyvitamin D-1alpha-hydroxylase in dysgerminomas: a novel form of humoral hypercalcemia of malignancy. Am J Pathol 165: 807-813.
14. Mudde AH, van den Berg H, Boshuis PG, et al, 1987 Ectopic production of 1,25-dihydroxyvitamin D by B-cell lymphoma as a cause of hypercalcemia. Cancer 59: 1543-1546.
15. Eid W, Wheeler TM, Sharma MD, 2004 Recurrent hypercalcemia due to ectopic production of parathyroid hormone-related protein and intact parathyroid hormone in a single patient with multiple malignancies. Endocr Pract 10: 125-128.
16. Mahoney EJ, Monchik JM, Donatini G, De Lellis R, 2006 Life-threatening hypercalcemia from a hepatocellular carcinoma secreting intact parathyroid hormone: localization by sestamibi single-photon emission computed tomographic imaging. Endocr Pract 12: 302-306.
17. LeGrand SB, Leskuski D, Zama I, 2008 Narrative review: furosemide for hypercalcemia: an unproven yet common practice. Ann Intern Med 149: 259-263.
18. Hamdy NA, Papapoulos SE, 2002 Management of malignancy-associated hypercalcaemia. Clinical Reviews in Bone and Mineral Metabolism 1: 65-76.
19. Papapoulos SE, 2008 Bisphosphonates: how do they work? Best Pract Res Clin Endocrinol Metab 22: 831-847.
20. Lacy MQ, Dispenzieri A, Gertz MA, et al, 2006 Mayo clinic consensus statement for the use of bisphosphonates in multiple myeloma. Mayo Clin Proc 81: 1047-1053.
21. Pecherstorfer M, Steinhauer EU, Rizzoli R, Wetterwald M, Bergstrom B, 2003 Efficacy and safety of ibandronate in the treatment of hypercalcemia of malignancy: a randomized multicentric comparison to pamidronate. Support Care Cancer 11: 539-547.
22. Shah S, Hardy J, Rees E, et al, 2002 Is there a dose response relationship for clodronate in the treatment of tumour induced hypercalcaemia? Br J Cancer 86: 1235-1237.
23. Veri A, D’Andrea MR, Bonginelli P, Gasparini G, 2007 Clinical usefulness of bisphosphonates in oncology: treatment of bone metastases, antitumoral activity and effect on bone resorption markers. Int J Biol Markers 22: 24-33.
24. Major P, Lortholary A, Hon J, et al, 2001 Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol 19: 558-567.
25. Papapoulos SE, Harinck HI, Bijvoet OL, Gleed JH, Fraher LJ, O’Riordan JL, 1986 Effects of decreasing serum calcium on circulating parathyroid hormone and vitamin D metabolites in normocalcaemic and hypercalcaemic patients treated with APD. Bone Miner 1: 69-78.
26. Adams JS, Hewison M 2006 Hypercalcemia caused by granuloma-forming disorders. In: Favus MJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 200-202.
27. Adams JS, Diz MM, Sharma OP, 1989 Effective reduction in the serum 1,25-dihydroxyvitamin D and calcium concentration in sarcoidosis-associated hypercalcemia with short-course chloroquine therapy. Ann Intern Med 111: 437-438.
28. Adams JS, Sharma OP, Diz MM, Endres DB, 1990 Ketoconazole decreases the serum 1,25-dihydroxyvitamin D and calcium concentration in sarcoidosis-associated hypercalcemia. J Clin Endocrinol Metab 70: 1090-1095.
29. Lipton A, Steger GG, Figueroa J, et al, 2008 Extended efficacy and safety of denosumab in breast cancer patients with bone metastases not receiving prior bisphosphonate therapy. Clin Cancer Res 14: 6690-6696.
30. Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR, 2001 The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res 16: 348-360.
31. Iguchi H, Aramaki Y, Maruta S, Takiguchi S, 2006 Effects of anti-parathyroid hormone-related protein monoclonal antibody and osteoprotegerin on PTHrP-producing tumor-induced cachexia in nude mice. J Bone Miner Metab 24: 16-19.
32. Sato K, Onuma E, Yocum RC, Ogata E, 2003 Treatment of malignancy-associated hypercalcemia and cachexia with humanized anti-parathyroid hormone-related protein antibody. Semin Oncol 30: 167-173.
33. Bilezikian JP, Silverberg SJ 2006 Primary Hyperparathyroidism. In: Favus MJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 181-185.
34. Lowe H, McMahon D, Walker M, Rubin M, Bilezikian J, Silverberg S, 2008 Normocalcemic Primary Hyperparathyroidism: Update on a New Clinical Phenotype. 30th American Society for Bone and Mineral Research Annual Meeting, 12-16 September, Montreal, Canada.
35. Bilezikian JP, Potts JT Jr., Fuleihan G, et al, 2002 Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century. J Clin Endocrinol Metab 87: 5353-5361.
36. Bilezikian JP, Khan AA, Potts JT Jr, Third International Workshop on the Management of Asymptomatic Primary Hyperthyroidism, 2009 Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 94: 335-339.
37. Farford B, Presutti RJ, Moraghan TJ, 2007 Nonsurgical management of primary hyperparathyroidism. Mayo Clin Proc 82: 351-355.
38. Hamdy NA, Gray RE, McCloskey E, et al, 1987 Clodronate in the medical management of hyperparathyroidism. Bone 8: Suppl 1: S69-S77.
39. Rossini M, Gatti D, Isaia G, Sartori L, Braga V, Adami S, 2001 Effects of oral alendronate in elderly patients with osteoporosis and mild primary hyperparathyroidism. J Bone Miner Res 16: 113-119.
40. Chow CC, Chan WB, Li JK, et al, 2003 Oral alendronate increases bone mineral density in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab 88: 581-587.
41. Parker CR, Blackwell PJ, Fairbairn KJ, Hosking DJ, 2002 Alendronate in the treatment of primary hyperparathyroid-related osteoporosis: a 2-year study. J Clin Endocrinol Metab 87: 4482-4489.
42. Makras P, Kaltsas GA, Athanasoulis T, et al, 2005 Treatment with oral bisphosphonates can increase the sensitivity of sestamibi radionuclide imaging in patients with primary hyperparathyroidism. Int J Clin Pharmacol Res 25: 19-28.
43. Greenspan SL, Holland S, Maitland-Ramsey L, et al, 1996 Alendronate stimulation of nocturnal parathyroid hormone secretion: a mechanism to explain the continued improvement in bone mineral density accompanying alendronate therapy. Proc Assoc Am Physicians 108: 230-238.
44. Khan AA, Bilezikian JP, Kung AW, et al, 2004 Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab 89: 3319-3325.
45. Reasner CA, Stone MD, Hosking DJ, Ballah A, Mundy GR, 1993 Acute changes in calcium homeostasis during treatment of primary hyperparathyroidism with risedronate. J Clin Endocrinol Metab 77: 1067-1071.
46. Lew JI, Solorzano CC, Irvin GL III, 2006 Long-term results of parathyroidectomy for hypercalcemic crisis. Arch Surg 141: 696-699.
47. Rubin MR, Lee KH, McMahon DJ, Silverberg SJ, 2003 Raloxifene lowers serum calcium and markers of bone turnover in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab 88: 1174-1178.
48. Favus MJ, Bushinsky D, Lemann JJ 2006 Regulation of calcium, magnesium, and phosphate metabolism. In: Favus MJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 76-83.
49. Shoback DM, Bilezikian JP, Turner SA, McCary LC, Guo MD, Peacock M, 2003 The calcimimetic cinacalcet normalizes serum calcium in subjects with primary hyperparathyroidism. J Clin Endocrinol Metab 88: 5644-5649.
50. Nemeth EF, Heaton WH, Miller M, et al, 2004 Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther 308: 627-635.
51. Cunningham J, Danese M, Olson K, Klassen P, Chertow GM, 2005 Effects of the calcimimetic cinacalcet HCl on cardiovascular disease, fracture, and health-related quality of life in secondary hyperparathyroidism. Kidney Int 68: 1793-1800.
52. Moe SM, Chertow GM, Coburn JW, et al, 2005 Achieving NKF-K/DOQI bone metabolism and disease treatment goals with cinacalcet HCl. Kidney Int 67: 760-771.
53. Peacock M, Bilezikian JP, Klassen PS, Guo MD, Turner SA, Shoback D, 2005 Cinacalcet hydrochloride maintains long-term normocalcemia in patients with primary hyperparathyroidism. J Clin Endocrinol Metab 90: 135-141.
54. Peacock M, Scumpia S, Bolognese MA, et al, 2003 Long-term control of primary hyperparathyroidism with cinacalcet HL (AMG 073). J Bone Miner Res 18: Suppl: 17.
55. Silverberg SJ, Rubin MR, Faiman C, et al, 2007 Cinacalcet hydrochloride reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab 92: 3803-3808.
56. Faggiano A, Tavares LB, Tauchmanova L, et al, 2008 Effect of treatment with depot somatostatin analogue octreotide on primary hyperparathyroidism (PHP) in multiple endocrine neoplasia type 1 (MEN1) patients. Clin Endocrinol (Oxf) 69: 756-762.
57. Marx SJ 2006 Familial hypocalciuric hypercalcemia. In: Favus MJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 188-190.
58. Brown EM, MacLeod RJ, 2001 Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81: 239-297.
59. Egbuna OI, Brown EM, 2008 Hypercalcaemic and hypocalcaemic conditions due to calcium-sensing receptor mutations. Best Pract Res Clin Rheumatol 22: 129-148.
60. Festen-Spanjer B, Haring CM, Koster JB, Mudde AH, 2008 Correction of hypercalcaemia by cinacalcet in familial hypocalciuric hypercalcaemia. Clin Endocrinol (Oxf) 68: 324-325.
61. Timmers HJ, Karperien M, Hamdy NA, de Boer H, Hermus AR, 2006 Normalization of serum calcium by cinacalcet in a patient with hypercalcaemia due to a de novo inactivating mutation of the calcium-sensing receptor. J Intern Med 260: 177-182.
62. Papapoulos SE, Clemens TL, Fraher LJ, Lewin IG, Sandler LM, O’Riordan JL, 1979 1, 25-dihydroxycholecalciferol in the pathogenesis of the hypercalcaemia of sarcoidosis. Lancet 1: 627-630.
63. Wysolmerski JJ 2006 Miscellaneous causes of hypercalcemia. In: Favus MJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 203-208.
64. Holick MF, 2007 Vitamin D deficiency. N Engl J Med 357: 266-81.
65. Lips P, van Schoor NM, Bravenboer N 2008 Vitamin D-related Disorders. In: Rosen CJ, (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, American Society for Bone and Mineral Research, USA; pp, 329-335.
Address for correspondence:
Polyzois Makras, Dept. of Endocrinology & Diabetes
251 Hellenic Air Force & VA General Hospital, 3 Kanellopoulou St., 115 25 Goudi, Athens,
Tel.: +30 210 7463606, Fax: +30 210 7715690 E-mail: makras@internet.gr
Received 22-01-09, Revised 28-02-09, Accepted 15-03-09