Section for Preventive Medicine, Department of Nephrology and General Medicine, University Medical Center, Albert-Ludwigs-University of Freiburg, Freiburg, Germany
Pheochromocytomas and Paragangliomas (PGL) form the group of paraganglial tumours which can occur in any paraganglia from the skull base to the pelvic floor. The terminology is not uniform. While the World Health Organization (WHO) applies pheochromocytoma exclusively to adrenal tumours, many clinicians use the term pheochromocytoma also for extra-adrenal abdominal and thoracic tumours, since by tradition pheochromocytoma is a vasoactive tumour. In contrast, head and neck paraganglioma is mostly only a space-occupying mass. The diagnosis is confirmed by both biochemical testing and radiological imaging. One third of patients with pheochromocytomas and paragangliomas are carriers of germline mutations in one of 6 genes and thus have a hereditary disorder. About 1% of Neurofibromatosis (NF) 1 patients have pheochromocytomas. All pheochromocytoma patients with NF 1 also show cutaneous lesions. About 50% of MEN2 patients harbour pheochromocytoma. The dominant lesion in this entity is Medullary Thyroid Carcinoma (MTC) occurring in up to 100% of patients. Von Hippel-Lindau disease (VHL)is found in about 20% of patients in association with pheochromocytoma. VHL is classified as type 1 predominantly without and type 2 predominantly with pheochromocytoma. Other important components of VHL are hemangioblastomas of the eye and Central Nervous System (CNS), renal clear cell carcinoma, multiple pancreatic cysts and islet cell carcinoma. PGL syndromes have been genetically characterized as PGL 1, 3 and 4 and are caused by mutations in the succinate dehydrogenase (SDH) subunit D, C and B genes, respectively (SDHD, SDHC and SDHB). Paraganglioma syndromes include predisposition to paraganglial tumours in any location, whereas PGL 3 patients mostly show only head and neck paragangliomas. All syndromes associated with paraganglial tumours are autosomal dominantly transmitted, but patients with SDHD mutations develop tumours only if they inherit the mutation from the father. Familial paraganglial tumours are characterized by younger age at diagnosis and more frequently multifocal and extra-adrenal abdominal pheochromocytomas. Patients with PGL 4 and less frequently VHL, are particularly predisposed to malignant pheochromocytoma. Endoscopic surgery is the primary treatment for pheochromocytoma. For malignant cases, chemotherapeutic as well as radionuclear approaches are available. No specific treatment has been proposed for prevention of the disease in inherited disorders. Thus, early diagnosis and regular follow-up are the only means for a better outcome.
MEN2, Molecular genetics, Paraganglioma, Paraganglioma Syndrome, Pheochromocytoma, von Hippel-Lindau
INTRODUCTION
Paragangliomas (PGL) are tumours of the paraganglia, a collection of neuroendocrine tissues and small organs with a common neuroectodermal origin and histological structure. Paraganglia may be either part of the sympathetic or the parasympathetic vegetative nervous system. Sympathetic paraganglial tissue (chromaffin) is mainly located in the adrenal medulla, but also in prevertebral and paravertebral thoraco-abdominal and pelvic paraganglia or ganglia in ovary, testis, vagina, urethra, prostate, bladder or liver and the Zuckerkandl organ. The parasympathetic paraganglia (non-chromaffin) are usually located in the vicinity of major arteries and nerves: the most common is the carotid body tumour, followed by jugular, vagal, tympanic, pulmonary and aortic paragangliomas.1,2
We prefer to use the term pheochromocytoma for both adrenal and sympathetic paraganglia (chromaffin) derived extra-adrenal tumours, which are usually endocrinologically active. We describe as paraganglioma those tumours arising from parasympathetic paraganglia (non-chromaffin) located in the skull-base and neck region which are usually hormonally inactive.3 The terminology is not uniform, the World Health Organization (WHO) Tumour Classification defining as pheochromocytoma only chromaffin tumours arising from the adrenal medulla, while all other tumours (chromaffin and non-chromaffin origin) being classified as extra-adrenal paragangliomas.4
In this work a clinical view of pheochromocytoma with particular reference to the hereditary forms is presented.
PHEOCHROMOCYTOMA EPIDEMIOLOGY
Pheochromocytoma is a rare tumour with an approximate incidence of 2-8 cases per million per year, with the highest incidence occurring between 50-60 years, without gender difference.5 It is responsible for 0.2-0.4% of cases of arterial hypertension.5 The malignancy rate is variable, from 2.4-26%.6,7 There are no histological proofs of malignancy to date and the only accepted criterion is the presence of metastasis. Histological indices, such as the Thompson PASS (Pheochromocytoma of the Adrenal Gland Scaled Score) or other immunohistochemistry or gene expression profiles might suggest malignancy but are still not diagnostic.8-10 The distant metastases are usually of hematologic origin, mostly involving bone, liver and lung. Since metastasis represents the only accepted criterion for malignancy, it is important to distinguish between metastasis and multifocality which may occur, especially in inherited disorders.11-13
HEREDITARY PHEOCHROMOCYTOMA
Up to 25% of pheochromocytoma cases are hereditary.11,14 It is to be noted that the first described case of pheochromocytoma (1886) was recently proven to be a patient with MEN2.15,16
Six pheochromocytoma-associated syndromes have thus far been identified. The first to be described was neurofibromatosis type 1 syndrome (NF1) reported by Suzuki et al in 1910.17 The most famous are the multiple endocrine neoplasia type 2 (MEN2) and von Hippel-Lindau Syndrome (VHL).18 Recently, three new syndromes associated with pheochromocytoma have been identified: PGL syndrome types 1, 3 and 4,13,19-21 all following an autosomal dominant inheritance pattern. For PGL 1, maternal imprinting of the susceptibility gene has been proposed, thus implying that only a carrier inheriting the mutation from the father will develop the disease; an exception to this rule has been observed, but the case is unclear.22,23
The presence of several reports of familial non-syndromic cases indicates that the prevalance of inherited cases might be higher than currently recorded.24,25
A summary of pheochromocytoma presentation in the different syndromes is represented in Table 1 .
Neurofibromatosis type 1
The susceptibility gene, NF1, located on the long arm of chromosome 17 (17q11.2) with 57 coding exons, is the largest pheochromocytoma susceptibilty gene described so far.26 The diagnosis is clinical and is based on the presence of café-au-lait spots, fibromatous tumours of the skin, axillary freckling and Lisch nodules of the iris. About 1-3% of patients with NF1 develop a pheochromocytoma.27 The mean age of pheochromocytoma onset is 42 years; 84-95% of patients have solitary adrenal tumours, 5-10% have bilateral adrenal disease and 0-6% have extra-adrenal pheochromocytomas; malignant pheochromocytomas are present in 3-12% of the cases.28,29 There is no correlation between the mutation type and the pheochromocytoma behaviour.29
Multiple endocrine neoplasia type 2
Two subtypes of this syndrome are associated with pheochromocytoma, MEN2A and 2B.
MEN2 is characterized by Medullary Thyroid Carcinoma (MTC) present in >90% of cases; in the MEN2A 20% of cases are diagnosed with primary hyperparathyroidism, whilst MEN2B is characterized by ganglioneuromas of the mucosa and marfanoid habitus.
Activating mutations of the RET proto-oncogene are involved in the pheochromocytoma genesis. The RET gene lies on the long arm of chromosome 10 (10q11.2) and consists of 16 exons, of which exons 10, 11, 13 and 16 are mainly associated with pheochromocytoma. About 50% of MEN2A and 2B cases develop a pheochromocytoma, but the overall and age-related penetrance varies between different mutations.30,31 The pheochromocytoma is usually adrenal, frequently bilateral and rarely extra-adrenal or malignant.29
Von Hippel-Lindau Syndrome
We distinguish two major subgroups of VHL Syndrome: VHL type 1 mainly without and VHL type 2 mainly with pheochromocytoma presentation.32 The clinical features of the VHL syndrome include retinal (von Hippel) and cerebellar (Lindau) hemangioblastoma, as well as brainstem and spinal hemangioblastoma. They also include presence of renal cysts and renal cell carcinoma, pancreatic cysts and islet cell tumours, endolymphatic sac tumours, as well as cysts and cystadenomas of epididymis and broad ligament (Figure 1).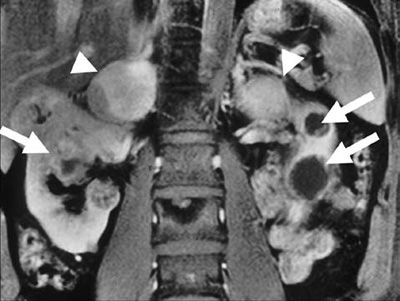
Figure 1. Sagittal MRI section shows bilateral cystic renal cell carcinoma (arrows) and bilateral adrenal pheochromocytoma (arrow heads) in a patient with von Hippel-Lindau disease.
The VHL gene lies on the short arm of chromosome 3 (3p25), with 3 exons coding for 2 isoforms of the protein.33 The mutations are spread in all three exons. Missense mutations, usually confer better prognosis and are more frequently detected in patients presenting with pheochromocytoma.34 About 20-30% of VHL type 2 patients develop a pheochromocytoma. The age at diagnosis is younger than in sporadic cases. They are frequently multiple: bilateral adrenal and multifocal extra-adrenal. Rarely, they are malignant.
Paraganglioma Syndromes
Three of the four described Paraganglioma Syndromes (PGL), Types 1, 3 and 4, are associated with pheochromocytoma. The susceptibility genes SDHB (PGL 4), SDHC (PGL 3) and SDHD (PGL 1) code the three subunits of the succinate dehydrogenase enzyme,35 while the fourth subunit coded by the SDHA gene is not associated with hereditary paraganglioma.36 Paraganglioma syndrome type 2, for which the susceptibility gene has not as yet been identified, has been diagnosed in families with head and neck paraganglioma, but not pheochromocytoma.37
Patients with SDHD gene (PGL 1) defect mainly develop head and neck paraganglioma rather than pheochromocytomas. The pheochromocytomas are usually benign and multifocal. One case with PGL 1 is represented in Figure 2.38 In contrast, patients with SDHB gene mutations (PGL 4) develop more frequently pheochromocytomas, single and extra-adrenal, with a higher percentage of malignant cases (up to 34%). For both syndromes the age at onset of tumour manifestations was earlier compared with the sporadic forms.12,39 Paraganglioma syndrome type 3 was initially associated with only head and neck paragangliomas, with no difference in age at onset as compared to the sporadic cases, and were mostly benign.40,41 Recently, two cases presenting with benign adrenal and extra-adrenal pheochromocytoma were described. Larger series are necessary for better definition of pheochromocytoma presentation within this syndrome.13,21
Other tumours that have been associated with paraganglioma syndromes are renal clear cell carcinoma (RCC) and Gastrointestinal Stromal Tumours (GIST). Whilst RCC is associated with mutations in the SDHB gene, mutation in all three genes have been identified in patients with the Carney-Stratakis dyad of paraganglioma and gastric stromal tumour.42,43
SYMPTOMS AND SIGNS
There is a long list of pheochromocytoma-associated symptoms and signs (Table 2 ) and they are all related to excessive catecholamine (epinephrine, norepinephrine and dopamine) excretion. The classical triad of palpitation, headache and excess sweatiness are present in only 15-24% of cases.44-46 In half of the patients, hypertension is present as a constant feature, whilst it is intermittent in the remaining half. Normal blood pressure is rare in pheochromocytoma patients, even though new studies have shown a lower prevalence (60-70%) of hypertension, especially in dopamine-secreting tumours.44-47
The symptoms of pheochromocytomas are usually intermittent due to the intermittent catecholamine excess. In 75% of cases the symptoms occur weekly, while in others this can be daily or at intervals of several months. Crisis due to untreated catecholamine excess can lead to heart insufficiency, pulmonary oedema, respiratory distress, heart arrhythmias, intracerebral bleeding and death.
Signs and symptoms of malignant pheochromocytoma are related to the infiltration of the tumours into both neighbourhood structures and distant organs.48
Asymptomatic pheochromocytoma is rare and it is mainly detected in mutation carriers after clinical screening. In these cases, retrospective analysis of symptoms and signs of the tumour manifestation suggests an asymptomatic or mildly symptomatic state.
Figure 2. Paraganglioma syndrome type 1. The left panel represents an 18F-DOPA image with at least 4 tumours identified. The right panel represents the MRI images of the 4 tumours: right carotid paraganglioma (A), left tympanic paraganglioma (B), large upper mediastinal (C) and small paratracheal paraganglioma (D).
DIAGNOSIS OF PHEOCHROMOCYTOMA
The diagnosis is based on biochemical testing and imaging. Biochemical testing alone is insufficient, especially in screening for tumours in patients at risk (mutation carriers).49,50
Biochemical Diagnosis
Excess of catecholamines (epinephrine, norepinephrine, dopamine) and their metabolites in the plasma or urine are the basis of biochemical testing. Due to intermittent catecholamine secretion, serial measurements might show different values. In the majority of symptomatic cases the catecholamine excess is also present in asymptomatic phases.
The most commonly applied tests include measurements of plasma and urinary epinephrine and norepinephrin, as well as the urinary vanillylmandelic acid (VMA) and the plasma free or urinary (fractioned or total) metanephrine and normetanephrine. Several methods have been used for this determination, including spectrophotometry, high pressure liquid chromatography (HPLC) and radioimmunoassays. The most sensitive method is the determination of plasma free metanephrines.51 The HPLC-Method used for the latter is available in only a limited number of medical centres, and thus the determination of plasma and urinary catecholamines and total urinary metanephrines are common practice. For urinary assays it might be important to concomitantly determine the creatinine clearance to detect eventual collection errors.
Usually at least a 2- to 3-fold level elevation of the assay’s given normal range is present in pheochromocytoma patients. False positive results may be present due to stress or concomitant use of drugs (acetaminophen, tricyclic antidepressants, phenoxybenzamine, L-DOPA, high-dose diuretics) or dietary products (caffein, nicotine) which might influence some of the assays or act on the physiology of catecholamines excretion.48
In screening of at risk patients (with known inherited disorder), the false negative results are much higher compared to the sporadic cases.51 One of the reasons could be the tumour size, being much smaller in routinely screened, at risk patients compared to symptomatic cases.52
Differences in the secretion patterns have been observed in the various localizations of the tumour as well between the tumours associated with different hereditary syndromes.52 Extra-adrenal pheochromocytomas as well as tumours associated with VHL syndrome, are mostly noradrenergic. Pheochromocytoma associated with MEN2 manifest more frequently an adrenergic pattern.
Radiological Imaging
Computer tomography (CT) and Magnetic Resonance Imaging (MRI) represent the methods of choice for cross-sectional imaging of pheochromocytoma. Using these methods, tumour of sizes 0.5 to 1 cm can be detected. Comparing the two methods, CT has the advantage of lower costs, but MRI has a slightly higher sensitivity and a lack of exposure to ionizing radiation, which is an important factor in hereditary cases undergoing continuous clinical follow-up.53
Since small extra-adrenal primary or metastatic tumours might be missed, anatomical imaging studies should be combined with functional (nuclear medicine) imaging studies for optimal results. The latter have a special role in localization of tumours in inherited disorders. They also give an idea of the endocrinological activity of the tumours, which can be useful in the further management (see below).
The three approaches include: 123I-MIBG Scinitgraphy, 18F-DOPA or 18F-DOPAMINE, positron emission tomography (PET) and 111In-octreotide scintigraphy (OCTREOSCAN).53 The radioactive tracer used in these approaches has an almost exclusively paraganglial tissue uptake.54,55 The most frequently used is 123I-MIBG-scintigraphy, widely available and highly specific (95-100%) but with a sensitivity of 77-90%.The uptake of 123I by the thyroid should be blocked by perchlorate administration. 18F-DOPAMINE or 18F-DOPA PET is in terms of sensitivity and specificity superior to the other two options; these can moreover be increased by prior carbidopa administration.56,57 Unfortunately, the latter is available only in a few highly specified centres and is thus currently not of use for routine clinical praxis. Although of low sensitivity, 111In-octreotide-scintigraphy may be useful in MIBG-negative, highly undifferentiated or hemorrhagic metastasis.
TREATMENT OF PHEOCHROMOCYTOMA
The goal is removal of the tumour. For effective blood pressure control and prevention of hypertensive crisis, a sufficiently long preoperative treatment (about 7 days) with alpha-blockers should be applied. The drug of choice is phenoxybenzamine, a non-specific and non-competitive irreversible alpha-blocker. The initial dose of 10 mg twice a day should be slowly increased (recommended increment: 10mg/die) under tight blood pressure measurement; usually a final dose of 1-2 mg/kg/die is necessary, given best in 3 doses.58 The aim is to achieve normotension, which should be documented with a 24h blood monitoring before the surgical intervention. The typical side effects of an effective alpha-blockade include tachycardia, orthostatic hypotension, gastrointestinal disturbances and oedema of the nasal mucosa. The last phenoxybenzamine dose should be given the night before surgery. To counterbalance the orthostatic hypotension induced by the alpha-receptors blockade, salt and liquid consumption should be advised. Alternatively, selective alpha 1-receptor blockers can be given; however, the lower effect on catecholamine excess along with a lack of studies on this medication render it inferior compared to phenoxybenzamine. Other drugs such as calcium channel blockers and beta-blockers as well as combined alpha and beta-blockers may be used.58 Calcium channel antagonists do not produce orthostatic hypotension and are thus very well tolerated in normotensive patients with paroxysmal hypertension. Beta-blockers are of interest in cases with tachycardia, but due to the potential hypertension caused by beta 2 receptor blockage, they should be administrated together with the alpha blockers.
Hypertensive crisis is managed with sodiumnitroprusside, urapidil or phentolamine. Intraoperative pressure elevations due to tumour manipulation can be well controlled by continuous sodiumnitroprusside infusions.
Benign Tumours
Surgical intervention should be performed only in specialized and experienced centres. Minimal invasive endoscopic surgery is the method of choice and should be considered as the gold standard. The procedure may be either laparoscopic or retroperineoscopic.59-62 Adrenal sparing surgery should be preferred to adrenalectomy, especially in hereditary cases where controlateral manifestations and frequent reintervention might occur.63,64 In the case of MEN2, the pheochromocytoma removal should precede the thyroid surgery. Extra-adrenal abdominal, thoracic and pelvic tumours may be removed via endoscopic procedure and, in the case of multifocal tumours, within one intervention.62,65 The catecholamine normalization should be documented after the operation. In the instance of bilateral adrenal surgery, even if adrenal sparing, an adrenal insufficiency should be excluded by an ACTH test.
Malignant Tumours
The treatment is not curative, its aim being to reduce the morbidity (symptoms) caused by catecholamine excess and tumour infiltration/compression.
Local malignant pheochromocytomas are primarily treated by surgery, if possible by organ preserving endoscopic surgery. Therapy with alpha antagonists should be considered in order to reduce symptoms caused by catecholamine excess.
If surgery is not possible, other treatment options include chemotherapy and MIBG therapy. Therapeutic administration of 131Iodine-metaiodobenzylguanidine (131I-MIBG) in MIBG-uptaking tumours (verified by diagnostic 123I-MIBG scan) is effective.66 Individual doses typically range from 3.7 to 7.4 GBq and will be repeatedly administered at intervals of several months. “High dose” MIBG therapy (270-700 MBq/kgKG, maximum 37 GBq) has been introduced by a San Francisco group, achieving 13% CR and 50% PR in 30 patients.67 The following dose dependent side effects have been observed: severe thrombocytopenia and neutropenia, hypothyroidism, hypertension, ovarian failure, nausea, vomiting, secondary infections. Conventional chemotherapy includes cyclophosphamide, vincristine and dacarbacine (“CVD”, Averbuch protocol) in 3 – 6 cycles depending on response.
The effect of other nuclear medicine treatments (90Yttrium- or 177Lutetium-DOTA-TOC/-NOC or -TATE) as well as chemotherapeutic drugs (Sorafenib, Sunitinib and VEGF antagonists) is currently under investigation.68,69
Hereditary Cases
At the moment there is no treatment which could prevent the tumour formation. The necessity of surgical intervention on clinically silent tumours identified within clinical screening of mutation carriers is still unclear. The wait-and-scan policy can be considered in pediatric patients, especially for syndromes related to benign manifestations. Each case should be assessed individually, taking into consideration concomitant “at risk” factors such as pregnancy or patient’s activity. Medical or nuclear treatment of hereditary cases does not differ from apparently sporadic cases.
FOLLOW-UP
After successful treatment, there is no recommendation for periodical follow-up for patients with apparently sporadic pheochromocytoma, without a germ-line mutation in one of the susceptibility genes.
Patients with a germ-line mutation (syndromic patients) as well as non-syndromic patients (in which no mutation has been found in the known susceptibility genes) but with specific features such as young age at presentation (<20), multiple tumours or family history for paraganglial tumours, should be monitored life-long for recurrence of the disease. In addition, syndromic patients should be clinically screened for other features associated with the specific syndrome (see above).
The screening should include both radiological imaging as well as biochemical tests. There is no universally accepted time interval in which the screening for pheochromocytoma recurrence is carried out. In our experience, for carriers of an SDHB mutation a yearly interval is recommended. The same interval is recommended for von Hippel-Lindau syndrome in which screening for tumours of the kidneys and the pancreas is to be included. Intervals of 2 to 3 years are probably sufficient for patients with mutations of the SDHC and SDHD gene.
Genotype specific recommendations within each syndrome are necessary for better management and counselling of the patients. Future discoveries in the genetics of these syndromes (e.g. modifier genes) will improve our approach and make it more specific for each case.
REFERENCES
1. Zak F, Lawson W 1982 Anatomy and topography. In The Paraganglionic Chemoreceptor System. Springer-Verlag.
2. Baysal BE, 2002 Hereditary paraganglioma targets diverse paraganglia. J Med Genet 39: 617-622.
3. Neumann H 2008 Pheochromocytoma. In Harrison’s Principles of Internal Medicine, The McGraw-Hill Companies, Inc; New York
4. DeLellis RA, Lloyd RV, Heitz PU, Eng C, 2004 World Health Organization (2004) WHO Classification of Tumours, Pathology and genetics of tumours of endocrine organs Lyon, IARC Press.
5. Yeo H, Roman S, 2005 Pheochromocytoma and functional paraganglioma. Curr Opin Oncol 17: 13-18.
6. Proye C, Vix M, Goropoulos A, Kerlo P, Lecomte-Houcke M, 1992 High incidence of malignant pheochromocytoma in a surgical unit. 26 cases out of 100 patients operated from 1971 to 1991. J Endocrinol Invest 15: 651-663.
7. Melicow MM, 1977 One hundred cases of pheochromocytoma (107 tumours) at the Columbia-Presbyterian Medical Center, 1926-1976: a clinicopathological analysis. Cancer 40: 1987-2004.
8. Thompson LD, 2002 Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 26: 551-566.
9. Thouennon E, Elkahloun AG, Guillemot J, et al, 2007 Identification of potential gene markers and insights into the pathophysiology of pheochromocytoma malignancy. J Clin Endocrinol Metab 92: 4865-4872.
10. McNicol AM, 2006 Histopathology and immunohistochemistry of adrenal medullary tumours and paragangliomas. Endocr Pathol 17: 329-336.
11. Neumann HP, Bausch B, McWhinney SR, et al, 2002 Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346: 1459-1466.
12. Neumann HP, Pawlu C, Peczkowska M, et al, 2004 Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations.[erratum appears in JAMA. 2004 Oct 13;292(14):1686]. JAMA 292: 943-951.
13. Peczkowska M, Cascon A, Prejbisz A, et al, 2008 Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab 4: 111-115.
14. Amar L, Bertherat J, Baudin E, et al, 2005 Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 23: 8812-8818.
15. Fr?nkel F, 1886 Ein Fall von doppelseitigem, v?llig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Ver?nderungen am Circulationsapparat und Retinits. Virchows Archiv f?r Pathologie, Anatomie und Physiologie 103: 244-263.
16. Neumann HP MD, Alexander Vortmeyer, Dieter Schmidt, et al, 2007 Evidence for MEN 2 in the Original Description of Classic Pheochromocytoma. N Engl J Med 357: 1311-1315.
17. Suzuki S, 1910 ?ber zwei Tumoren aus Nebennierenmarkgewebe. Berliner Klinische Wochenschrift 47: 1623.
18. Neumann HP, Berger DP, Sigmund G, et al, 1993 Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med 329: 1531-1538.
19. Baysal BE, Ferrell RE, Willett-Brozick JE, et al, 2000 Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287: 848-851.
20. Astuti D, Latif F, Dallol A, et al, 2001 Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69: 49-54.
21. Mannelli M, Ercolino T, Giache V, Simi L, Cirami C, Parenti G, 2007 Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet 44: 586-587.
22. Pigny P, Vincent A, Cardot Bauters C, et al, 2008 Paraganglioma after maternal transmission of a succinate dehydrogenase gene mutation. J Clin Endocrinol Metab 93: 1609-1615.
23. Neumann HP, Erlic Z, 2008 Maternal Transmission of Symptomatic Disease with SDHD Mutation: Fact or Fiction? J Clin Endocrinol Metab 93: 1573-1575.
24. Opocher G, Schiavi F, Iacobone M, et al, 2006 Familial nonsyndromic pheochromocytoma. Ann N Y Acad Sci 1073: 149-155.
25. Dahia PL, Hao K, Rogus J, et al, 2005 Novel pheochromocytoma susceptibility loci identified by integrative genomics. Cancer Res 65: 9651-9658.
26. Wallace MR, Marchuk DA, Andersen LB, et al, 1990 Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 249: 181-186.
27. Riccardi VM, 1981 Von Recklinghausen neurofibromatosis. N Engl J Med 305: 1617-1627.
28. Opocher G, Conton P, Schiavi F, Macino B, Mantero F, 2005 Pheochromocytoma in von Hippel-Lindau disease and neurofibromatosis type 1. Fam Cancer 4: 13-16.
29. Bausch B, Borozdin W, Neumann HPH, 2006 European-American Pheochromocytoma Study G. Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma. N Engl J Med 354: 2729-2731.
30. Machens A, Brauckhoff M, Holzhausen HJ, Thanh PN, Lehnert H, Dralle H, 2005 Codon-specific development of pheochromocytoma in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab 90: 3999-4003.
31. Quayle FJ, Fialkowski EA, Benveniste R, Moley JF, 2007 Pheochromocytoma penetrance varies by RET mutation in MEN 2A. Surgery 142: 800-805.
32. Neumann HP, Wiestler OD, 1991 Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus.[see comment]. Lancet 337: 1052-1054.
33. Latif F, Tory K, Gnarra J, et al, 1993 Identification of the von Hippel-Lindau disease tumor suppressor gene.[see comment]. Science 260: 1317-1320.
34. Maher ER, Webster AR, Richards FM, et al, 1996 Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet 33: 328-332.
35. Eng C, Kiuru M, Fernandez MJ, Aaltonen LA, 2003 A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer 3: 193-202.
36. Baysal BE, Lawrence EC, Ferrell RE, 2007 Sequence variation in human succinate dehydrogenase genes: evidence for long-term balancing selection on SDHA. BMC Biol 5: 12.
37. Mariman EC, van Beersum SE, Cremers CW, Struycken PM, Ropers HH, 1995 Fine mapping of a putatively imprinted gene for familial non-chromaffin paragangliomas to chromosome 11q13.1: evidence for genetic heterogeneity. Hum Genet 95: 56-62.
38. Reisch N, Walz MK, Erlic Z, Neumann HP, 2009 [Pheochromocytoma - still a challenge]. Der Internist 50:27-35
39. Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al, 2006 Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 91: 827-836.
40. Baysal BE, Willett-Brozick JE, Filho PA, Lawrence EC, Myers EN, Ferrell RE, 2004 An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J Med Genet 41: 703-709.
41. Schiavi F, Boedeker CC, Bausch B, et al, 2005 Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 294: 2057-2063.
42. Vanharanta S, Buchta M, McWhinney SR, et al, 2004 Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 2004;74: 153-159.
43. McWhinney SR, Pasini B, Stratakis CA, 2007 Familial gastrointestinal stromal tumours and germ-line mutations. N Engl J Med 357: 1054-1056.
44. Plouin PF, Degoulet P, Tugaye A, Ducrocq MB, Menard J, 1981 [Screening for phaeochromocytoma: in which hypertensive patients? A semiological study of 2585 patients, including 11 with phaeochromocytoma (author’s transl)]. La Nouvelle presse medicale 10: 869-872.
45. Baguet JP, Hammer L, Mazzuco TL, Chabre O, et al, 2004 Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol 150: 681-686.
46. Mannelli M, Ianni L, Cilotti A, Conti A, 1999 Pheochromocytoma in Italy: a multicentric retrospective study. Eur J Endocrinol 141: 619-624.
47. Proye C, Fossati P, Fontaine P, et al, 1986 Dopamine-secreting pheochromocytoma: an unrecognized entity? Classification of pheochromocytomas according to their type of secretion. Surgery 100:1154-1162.
48. Manger WM, Eisenhofer G, 2004 Pheochromocytoma: diagnosis and management update. Current Hypertens Rep 6: 477-484.
49. Young WF Jr 2008 Endocrine hypertension. In Williams Textbook of Endocrinology, Philadelphia, PA, Saunders Elsevier.
50. Mariola P?czkowska, Zoran Erlic, Michael M. Hoffmann, et al, Impact of Screening Kindreds for SDHD p.Cys11X as a Common Mutation Associated with Paraganglioma Syndrome Type 1. J Clin Endocrinol Metab 93: 4818-4825.
51. Lenders JW, Pacak K, Walther MM, et al, 2002 Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287: 1427-1434.
52. Eisenhofer G, Lenders JW, Goldstein DS, et al, 2005 Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem 51: 735-744.
53. Ilias I, Pacak K, 2004 Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab 89: 479-491.
54. van der Harst E, de Herder WW, Bruining HA, et al, 2001[(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in begnign and malignant pheochromocytomas. J Clin Endocrinol Metab 86: 685-693.
55. Furuta N, Kiyota H, Yoshigoe F, Hasegawa N, Ohishi Y, 1999 Diagnosis of pheochromocytoma using [123I]-compared with [131I]-metaiodobenzylguanidine scintigraphy. Int J Urol 6: 119-124.
56. Hoegerle S, Ghanem N, Altehoefer C, et al, 2003 18F-DOPA positron emission tomography for the detection of glomus tumours. Eur J Nucl Med Mol Imaging 230: 689-694.
57. Hoegerle S, Nitzsche E, Altehoefer C, et al, 2002 Pheochromocytomas: detection with 18F DOPA whole body PET--initial results. Radiology 222: 507-512.
58. Pacak K, 2007 Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab 92: 4069-4079.
59. Walz MK, Peitgen K, Walz MV, et al, 2001 Posterior retroperitoneoscopic adrenalectomy: lessons learned within five years. World J Surg 25: 728-734.
60. Walz MK, Petersenn S, Koch JA, Mann K, Neumann HP, Schmid KW, 2005 Endoscopic treatment of large primary adrenal tumours. Br J Surg 92: 719-723.
61. Walz MK, Alesina PF, Wenger FA, et al, 2006 Laparoscopic and Retroperitoneoscopic Treatment of Pheochromocytomas and Retroperitoneal Paragangliomas: Results of 161 Tumours in 126 Patients. World J Surg 30:1-10.
62. Barczynski M, Konturek A, Golkowski F, et al, 2007 Posterior retroperitoneoscopic adrenalectomy: a comparison between the initial experience in the invention phase and introductory phase of the new surgical technique. World J Surg 31: 65-71.
63. Yip L, Lee JE, Shapiro SE, et al, 2004 Surgical management of hereditary pheochromocytoma. J Am Coll Surg 198: 525-534
64. Walz MK, Peitgen K, Diesing D, et al, 2004 Partial versus total adrenalectomy by the posterior retroperitoneoscopic approach: early and long-term results of 325 consecutive procedures in primary adrenal neoplasias. World J Surg 28: 1323-1329.
65. Walz MK, Peitgen K, Neumann HP, Janssen OE, Philipp T, Mann K, 2002 Endoscopic treatment of solitary, bilateral, multiple, and recurrent pheochromocytomas and paragangliomas. World J Surg 26: 1005-1012.
66. Safford SD, Coleman RE, Gockerman JP, et al, 2003 Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery 134: 956-962.
67. Fitzgerald PA, Goldsby RE, Huberty JP, et al, 2006 Malignant pheochromocytomas and paragangliomas: a phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG). Ann NY Acad Sci 1073: 465-490.
68. Forrer F, Riedweg I, Maecke HR, Mueller-Brand J, 2008 Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q J Nucl Med Mol Imaging 52: 334-340.
69. Wehrmann C, Senftleben S, Zachert C, Muller D, Baum RP, 2007 Results of individual patient dosimetry in peptide receptor radionuclide therapy with 177Lu DOTA-TATE and 177Lu DOTA-NOC. Cancer Biother Radiopharm 22: 406-416.
70. Manger WM, Gifford RW, 2002 Pheochromocytoma. J Clin Hypertens 4: 62-72.
Address for correspondence:
Prof. Dr. Hartmut P.H. Neumann, Medizinische Universit?tsklinik, Hugstetter Str. 55,
D-79106 Freiburg i.Br., Tel.: +49 761 270 3578, Fax: +49 761 270 3778,
e-mail: hartmut.neumann@uniklinik-freiburg.de
Received 01-11-08, Revised 30-11-08, Accepted 10-12-08