1Department of Neurosurgery, Toronto Western Hospital, University of Toronto, Toronto, Ontario, Canada,
2&3 Department of Laboratory Medicine, St. Michael Hospital, University of Toronto, Toronto, Ontario, Canada
Pituitary hyperplasia is rare, difficult to diagnose and sometimes controversial. The hyperplasia could be physiologic which is usually reversible, or pathologic which varies in presentation from incidental to tumor like lesion with and without hormonal disturbance. Any pituitary cell is capable of undergoing hyperplasia in the presence of the right stimuli. In this article we summarize the various pathologic and morphologic features of each subtype of pituitary hyperplasia, give an account of the molecular, hormonal and cellular basis of this condition and outline its clinical significance, differential diagnosis and prognosis.
Human pituitary, hyperplasia, morphology, basis, immunohistochemistry
INTRODUCTION
Pituitary hyperplasia has been known for a long period of time and yet remains poorly understood. It is an uncommon condition that is inadequately studied, difficult to diagnose and under-diagnosed 1,2. The aim of this review is to shed light on this condition and to discuss it based on the knowledge of the past aided by the recent advances in the fields of radiology, pathology and cellular and molecular biology.
DEFINITION
Pituitary hyperplasia is defined as a non-neoplastic increase in one or more functionally distinct types of pituitary cells2,3. The acini, though expanded, remain intact1,3,4. This numerical increase in the number of the pituitary cells may or may not be abnormal, and may or may not be associated with increase in the quantity of the product of the hyperplastic cells, but does not include change in their quality. A good example of normal physiological hyperplasia associated with increase in the quantity of the product of the hyperplastic cells is that of the prolactin cells during pregnancy, which always reverses itself after pregnancy ends. At the other extreme, some forms of pituitary hyperplasia (e.g. those of thyrotroph) could be large enough to compress adjacent structures and mimic neoplastic process5-8.
The hyperplastic cells by definition are polyclonal in nature2. In hyperplasia there is an increase in protein synthesis, total protein content, thymidine or bromodeoxyuridine incorporation, DNA content and cell number but not in the cell size or protein: DNA ratio9.
MOLECULAR BASIS OF PITUTARY HYPERPLASIA
Understanding pituitary hyperplasia requires an understanding of the molecular and cellular detail of the processes of cell hyperplasia in general. Though a detailed account of this is beyond the scope of this review, a brief discussion follows.
One basic fact to start with is that for pituitary cell hyperplasia to occur a stimulus must have happened initiating these cells to respond. This stimulus can be physiological or pathological.
Pituitary cell hyperplasia represents one of several different ways by which cells respond to stimuli. These responses include hyperplasia, hypertrophy, atrophy and metaplasia (Figure1). The stimulus may be an increase in demand like pregnancy, hormonal change like increased CRH post adrenalectomy or other stimuli like viral infection or exposure to chemicals, etc. Though the stimulus and the response may be physiologically appropriate, it is probably the type, the magnitude and the duration of the stimulus that determine the extent of pituitary hyperplasia. A good example of this relationship between the stimulus and the response of the pituitary cells is the large compressive and occasionally irreversible thyrotroph hyperplasia resultant of prolonged primary hypothyroidism, a response that might have been appropriate and reversible in cases of temporary primary hypothyroidism.
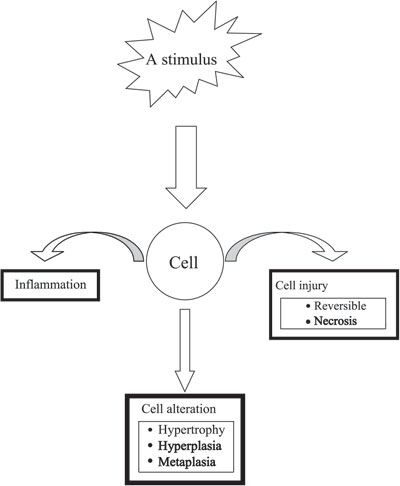
Figure 1. Illustration of deferent responses of a pituitary cell to a stimulus.
No matter what the type of stimulus and what the cell type receiving it, it is assumed that the basis of the interaction in cases of hyperplasia is at the level of the cell cycle. The cell cycle is a series of events undergone by the hyperplastic cells. G0 refers to the resting state, followed by G1, a phase of cell growth and enlargement. This phase is then followed by S phase in which the cell replicates its DNA and enters into G2 phase, which is a second but shorter growth phase. Finally, this ends by cell division in the M phase. The rate of progression through the cell cycle is controlled by cell cycle kinases. Each phase of the cell cycle is regulated by a specific kinase9. Sufficient kinase activity needs to be present for the cell to proceed through the cell cycle. Thus, the presence of factors that activate these kinases (e.g p21) or others that inhibit them (e.g. p27 and p57) can determine the way the cell proceeds through the cell cycle4,9-12 (Figure 2).
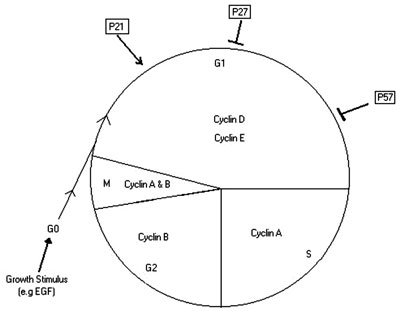
Figure 2. Illustration of the Control of cell cycle in pituitary cell.
The stimulus results in expression of growth factors that mediate their effect through interaction with specific receptors leading to expression of genes that promote DNA synthesis and entry of the cells into the cell cycle. Similar interaction is assumed to occur between growth factors and their receptors leading to a decrease in the rate of apoptosis in tissues undergoing hyperplasia13-15. The role of the receptors' stimulation or blockade in pituitary cell proliferation is well illustrated by N-Methyl-D-Aspartate (NMDA), dopamine (D-2), cannabinoid CB1 and leptin receptors16-21. For example, NMDA receptor activation inhibits cell proliferation and promotes cell death, while its blockade promotes cells to go through the cell cycle16-18. The growth factors and other factors that influence the pituitary cell cycle are numerous and are likely to interact with many others in a complex way. They include proliferative ones like epidermal growth factor (EGF), transforming growth factor-a (TGF-a), vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), nerve growth factor (NGF), insulin-like growth factor-1 (IGF-1), and the monoamine neurotransmitters; also antiproliferative ones like transforming growth factor-b (TGF-b), glutamate, gamma-aminobutyric acid, dopamine agonists and opioid peptides2,4,10,12,13,22-26. EGF, for example, causes activation of cell cycle kinases, phosphorylation of pRB and increase in thymidine incorporation, thus driving the cells through the cell cycle and thereby leading to hyperplasia9.
Theoretically the derangement could be at the level of the stimulus by being sustained, abnormal or amplified; or at the level of the receptors by being up or downregulated and hence allowing the cells to replicate and/or not go into apoptosis.
The balance between cell replication and cell death is essential in the control of the size and shape of every mature organ of the body (Figure 3). Growth and antigrowth factors are central in controlling these responses and an imbalance between these two processes is fundamental in the development of hyperplasia. The rate of cell death and cell replication differs considerably between different mature tissues, and while it is high for both processes in the gastrointestinal tract, it is low for both processes in the pituitary gland13,27. Following exposure to stimuli, hyperplasia can be induced in the target organ by increase in the rate in the cell replication as well as by suppression of the apoptosis at the same time. In the pars distalis, however, the apoptosis is a rare event and its role in pituitary hyperplasia is not clear2.

Figure 3. Illustration of the role of the imbalance between cell proliferation and apoptosis in the formation of pituitary hyperplasia.
HORMONAL INFLUENCE ON PITUITARY HYPERPLASIA
Pituitary cell proliferation is governed by numerous hormonal factors that include hypothalamic growth hormone releasing hormones, lack of dopaminergic, inhibition of lactotrophs and lack of feedback suppression by adrenal, gonadal or thyroid hormones.
Growth hormone releasing hormone (GHRH) can cause somatotroph proliferation leading to hyperplasia2,22,28-30. GHRH effect is probably mediated through cyclic AMP (cAMP)4,31. Corticotropin-releasing hormone (CRH) excess can lead to corticotroph hyperplasia, which has been documented in Cushing's and Addison's diseases2,4,22,32,33. Vasopressin (VP) may also be an important hormonal factor in corticotroph hyperplasia4. Thyrotropin releasing hormone (TRH) is likely an important cause of thyrotroph and lactotroph hyperplasia linked to primary hypothyroidism6,7,22,34-43. Gonadotrophin releasing hormone (GnRH) has also been implicated as a cause of hyperplasia in clinically documented cases of hypergonadotropic ovarian failure with pituitary hyperplasia4,44,45. Pituitary hyperplasia has also been documented after gonadorelin (GnRH-analogue) therapy and in patients with gonadal dysgenesis46,47.
Estrogen excess for a prolonged period of time has been documented as inducing pituitary hyperplasia both clinically, as in prolonged treatment of Turner's syndrome, and in animal models4,17,20,48-50.
Loss of inhibitory hormones also plays a role in the pituitary hyperplasia pathogenesis. This is well illustrated by lactotroph hyperplasia induced by elimination of D2 receptor activity due to dopamine's lack of inhibitory effect 4,20-22,38. It has been suggested that estrogen induces pituitary hyperplasia by antagonizing the antiproliferative effect of dopamine on the pituitary cells 51. Somatostatin, glucocorticoid, thyroid hormones as well as the deficiency of gonadal steroids may also be important players in pituitary hyperplasia pathogenesis4,22,52. For example, thyroid hormone and estradiol are known to inhibit estrogen induced pituitary hyperplasia in animal models53.
Other hormonal and growth factors that could play a role in the pathogenesis of pituitary hyperplasia include vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating peptide, as well as platelet-derived growth factor and ciliary neurotrophic factor22,25.
CELLULAR BASIS OF PITUITARY HYPERPLASIA
Following the discussion of the molecular and hormonal basis of pituitary hyperplasia, we will now discuss it on a cellular level.
We start this by a quick look at the anatomy and histology of the pituitary gland. In the large majority of mammalian species, the pituitary gland consists of three anatomically distinct zones: the anterior, intermediate and neural lobes. A separate intermediate lobe is lacking in the human gland, giving rise to the misconception that in humans this lobe is vestigial and largely consists of the cystic structures between the anterior and posterior lobes. The fact is that the cystic structures are chiefly Rathke's pouch remnants, some of which may grow into larger cysts. What really happens is that, at the end of fetal development, the pituitary cleft becomes obliterated providing physical contact between the anterior and intermediate lobes. The cells of the latter migrate and become largely incorporated within the anterior lobe. This means that the glandular part of the human pituitary includes both anterior and intermediate lobes; therefore, it should preferably be called pars distalis54. The pars distalis is composed of relatively uniform-size acini populated by functionally distinct cell types. A delicate, capillary rich stromal network, best seen on silver preparations, surrounds each acinus. The integrity of this stromal network is a major distinctive feature between pituitary hyperplasia and adenoma. The five major pars distalis cell types are: (1) the corticotrophs which are middle-sized, ovoid, PAS positive cells and produce the adrenocorticotropic hormone (ACTH), a-melanocyte stimulating hormone (a-MSH), lipotrophin (LPH) and endorphins all of which are derived from the precursor polypeptide proopiomelanocortin (POMC),
(2) the thyrotrophs which are angular shaped, moderately PAS positive and produce thyroid-stimulating hormone (TSH), (3) the gonadotrophs which are medium-sized, ovoid, basophilic, PAS positive and produce both follicle-stimulating hormone (FSH) and leutinizing hormone (LH), (4) the somatotrophs which are ovoid-polygonal acidophilic cells and produce growth hormone (GH), and (5) the lactotrophs which are small chromophobes with acidophilic variants and produce prolactin (PRL). There are also the mammosomatotrophs, which contain both GH and PRL, and the pars intermedia-derived proopiomelanocortin-producing cells (POMC cells). The cytodifferentiation of these cells in early development depends on several factors that include transcription factors like Pit-1, hypothalamic releasing factors like GHRH, growth factors like bFGF and cytokines like interleukin-2 (IL-2) and IL-64.
The spatial and proportional representation of the various cell types in the anterior pituitary gland is fairly constant, though the ratios of different pituitary cells may differ between sexes and according to age and the physiologic state, such as pregnancy or target organ failure2,4. In human adults, somatotrophs account for approximately 50% of cells in the anterior pituitary gland, gonadotrophs 15-20%, corticotrophs 10%, and thyrotrophs 5%. Lactotrophs account for 9% of the cells in the adult male and nulliparous female, but up to 31% in the multiparous female indicating that pregnancy-induced lactotroph hyperplasia does not completely reverse55.
The pituitary cells are not randomly distributed through the gland1-4, the corticotrophs being concentrated mainly in the median wedge. The somatotrophs occupy most of the lateral wings with the exception of a narrow posterolateral area. Many are also scattered in the median wedge. The lactotrophs are scattered fairly evenly over the pars distalis, though are more prominent at the posterolateral rim of the lateral wings. Thyrotrophs reside chiefly within the anterior median wedge with a minor extension into the lateral wings. The gonadotrophs are evenly distributed through the pars distalis.
The human pituitary gland increases in weight from 100 mg at birth to the adult weight of 600 mg4. This weight gain is accompanied by a several-fold increase in cell number indicating that both proliferation and hypertrophy of pituitary cells take place postnatally4. Though stem cells are a possible source of the new cells, both proliferation and transdifferentiation of mature differentiated cells have been demonstrated1,4,56,57.
The overwhelming evidence indicates that hyperplasia stems from proliferation of different pituitary cells4. There is, however, evidence, to indicate that some of the cells in the hyperplastic gland originate from transdifferentiation of other cell types4,57,58. Transdifferentiation is best accounted for by the existence of bi-hormonal cells; mammosomatotrophs (produ-cing GH and PRL) and thyrosomatotrophs (produ-cing GH and TSH). In both these examples, cells from one cell type are recruited in the formation of cells of the other hyperplastic type, an event that reverses once the stimulus is over (reversible transdifferentiation)1,57,58.
MORPHOLOGY OF PITUITARY HYPERPLASIA
Pituitary hyperplasia is not a single morphologic entity. It is rather a process encompassing several phases, ranging from the slight increase in the number of normal cells not affecting tissue architecture to massive space-occupying enlargement of the gland involving significant alteration in both tissue architecture and morphology1. Pituitary hyperplasia can be diffuse or nodular, unifocal or multifocal1-4. The diffuse type signifies numerical increase in the pituitary cells without major alteration in the cell morphology or the acinar architecture1.When diffuse pituitary hyperplasia is marked, the acini become slightly but evenly enlarged. This morphological type can be difficult to diagnose in a fragmented specimen.
Focal pituitary hyperplasia signifies a small, circumscribed increase of pituitary cell type. It is usually an autopsy finding and with no clinical significance1. Nodular pituitary hyperplasia is the more advanced focal type. The affected acini in the nodular type are variably enlarged and populated by an increased number of the affected cell type whose morphology is more homogenous as compared to the diffuse type. The affected cells may be enlarged and there might be a decrease in the number of unaffected cell types in the same acini. Because pituitary adenoma is a major pathological differential diagnosis for nodular hyperplasia, it is important to know some of the key features that distinguish nodular hyperplasia as compared to pituitary adenoma, including: lack of disruption of the reticulin network, though this might happen focally in severe forms; hyperplastic cell mass, which is never monomorphic; indistinctly demarcated area of hyperplasia; and non-evident compression of adjacent structures and formation of pseudocapsule. The nodular type may be focal or multinodular throughout the gland. The underlying cause(s) of these different forms of hyperplasia are unknown but may reflect the influence of the local environment4.
DIAGNOSIS OF PITUITARY HYPERPLASIA
Diagnosis of pituitary hyperplasia is difficult. This is due to several reasons which include the fact that pituitary cell numbers vary depending on the age, sex and physiologic state. Also complicating the issue are the following characteristics: normal pituitary cell counts are not firmly established for most cell types; morphologic criteria are lacking; the regional distribution of several pituitary cells is difficult to incorporate in fragmented surgical specimens or those taken from a non-specified anatomic site; and, last but not least, issues arise related to the quality of the surgical specimen (e.g. if it is crushed) leading to distortion of the architecture and cell membrane lysis resulting in leak of content and hence hindering the reliability of the immunostains1,2,59. Furthermore, some pituitary cells (e.g. corticotrophs) normally tend to aggregate thus expanding acini in a manner simulating hyperplasia2.
Despite the recent advances in the field of pathology, the diagnosis of pituitary hyperplasia remainsv dependent on the classic techniques such as histochemical stains (reticulin, PAS) and immunostains for the pituitary hormones.
The proliferation markers are not very useful in the diagnosis of pituitary hyperplasia. For example, MIB-1 index in pituitary hyperplasia, though found to be higher than normal, was still too low to be used as reliable marker for cell proliferation1,2. Similarly, the molecular techniques such as in situ hybridization, polymerase chain reaction and blotting studies, have contributed little to the diagnosis of pituitary hyperplasia2.
HYPERPLASIA OF DIFFERENT CELL TYPES
Any cell type in the pars distalis can give rise to hyperplasia1,2. There is, however, a difference in the etiology, pathogenesis, pathology, frequency, extent and clinical significance. Usually only one cell type increases in number hence forming pituitary hyperplasia. Occasionally, however, more than one cell type can become hyperplastic28,38,47. This latter situation is exemplified by hyperplasia of the thyrotrophs and lactotrophs in the setting of prolonged primary hypothyroidism as will be seen from the discussion below.
Corticotroph hyperplasia
Corticotroph hyperplasia occurs in long-standing Addison's disease, cases of ectopic overproduction of CRH and the idiopathic form in some cases of ACTH-dependant Cushing's disease1,2,4,32,56. Morphologically corticotroph hyperplasia is nearly always nodular1,2. The hyperplastic cells are smaller than normal, amphophilic, PAS-positive and may display mild Crook's hyaline changes1,2. In rats, Post bilateral adrenalectomy, there is an initial decline in the number of corticotrophs, which may be artifactual due to degranulation resulting from ACTH release. This is followed by regranulation and increase in both cell sizes and numbers4. The possible mechanisms are proliferation of the corticotrophs and proliferation and differentiation of uncommitted cells. Transdifferentiation from other differentiated cells though possible is less likely since the corticotroph upstream transcription element (CUTE) is corticotroph specific4.
Pars intermedia (PI) derived POMC-cells can also undergo hyperplasia, which is likely to occur close to the anterior rim, at the median wedge junction with the lateral wings and in the posterior part of the median wedge. It is also well represented by the well-known basophil invasion within the posterior lobe1,2,54. PI derived POMC-cells hyperplasia is usually an incidental finding that is common in old age and may give rise to tumors (the two types of silent corticotroph adenomas), but pathologists may not realize its separate identity and misdiagnose it as corticotroph hyperplasia54.
Somatotroph hyperplasia
Somatotroph hyperplasia is not recognized in any physiological situation4. Though rare, it has, however, been demonstrated in cases of ectopic GHRH excess such as ectopic production from pheochromocytoma or various carcinoid tumors. Hypothalamic GHRH excess due to hypothalamic gangliocytoma or hypothalamic regulatory defect as in cases of congenital gigantism is extremely rare1,4,28,29. The hyperplastic cells are strongly acidophilic and GH-immunopostive, though some may also be PRL-immunopostive. Somatotroph hyperplasia is mostly of the diffuse morphologic type.
Mammosomatotroph hyperplasia is very rare and is associated with congenital gigantism and McCune-Albright syndrome1,2.
Lactotroph hyperplasia
Lactotroph hyperplasia is the most common type and is a well-recognized physiological response in pregnancy and lactation1,2,4,55,56. At term, around 70 % of the pars distalis cells may be PRL-immunopostive1. Gestational lactotroph hyperplasia is of the diffuse morphologic type and reverses following the end of gestation and lactation1,2. There is, however, a proportion of lactotrophs that may not reverse completely leading to a higher proportion of lactotrophs in the pituitaries of multiparous compared to nulliparous women1,2,55. This may come from maturation of uncommitted cells, lactotroph proliferation and also from transdifferentiation of other cells, particularly somatotrophs and mammosomatotrophs4,15,55,58.
Lactotroph hyperplasia occurs with exogenous estrogen administration, pituitary space-occupying lesion due to stalk section effect, resulting in loss of dopaminergic inhibition on the lactotrophs, in conjunction with corticotroph adenoma probably due to increased endorphin production, and finally in some patients with prolonged primary hypothyroidism, probably as a consequence of elevated TRH1,2,4,38. Idiopathic lactotroph hyperplasia is very rare but well documented and may lead to hyperprolactinemia1,2,56,60.
Non-gestational lactotroph hyperplasia can morphologically be either nodular, diffuse or, rarely, take both forms in the same patient1,2. The hyperplastic cells are large with abundant chromophobic to slightly acidophilic cytoplasm that is PAS-negative2. The hyperplastic cells come from lactotrophs and recruited (trandifferentiated) somatotrophs.
Gonadotroph hyperplasia
Gonadotroph hyperplasia is a rare but well recognized response to castration in animal models and in humans with long-standing primary hypogonadism, Turner's or Klinefelter's syndromes1,2,4. It has also been reported in cases of secondary hypergonadotropic ovarian failure44,45. Morphologically gonadotrophs hyperplasia tends to be of the diffuse type1,2.The hyperplastic cells are large, ovoid and vaculated with an eccentric nucleus and dilated RER giving the signet-ring appearance1,2. It is more difficult to make the diagnosis of gonadotroph hyperplasia than other types of pituitary hyperplasia due to reasons that include their scattered distribution in the pars distalis, the wide range of normal gonadotroph content and variation due to age and sex1,2.
Thyrotroph hyperplasia
Many cases of thyrotroph hyperplasia resulting from prolonged primary hypothyroidism have been reported2,4,6,7,34,35,37-43,61-64. This response is usually reversible with the institution of thyroxine replacement therapy1,2,6,7,35,37,42,43,62. TRH is likely a major player initiating both thyrotroph proliferation and possibly transdifferentiation from lactotrophs and somatotrophs4,57. The hyperplastic cells are large, ovoid, pale with vaculated chromophobic to slightly acidophilic cytoplasm with several lysosomes and variable TSH-immunoreactivity1,2,7. Morphologically most thyrotroph hyperplasia are of the nodular type but can be diffuse or multifocal1,2. Thyrotroph hyperplasia can be large enough to mimic tumor and compress the adjacent structures, a fact that needs to be kept in mind in order to avoid confusion with pituitary adenoma1,2,6,7,36,41,43,62,65. This misdiagnosis is more likely to arise if there is an associated hyperprolactinemia since the pituitary lesion might be interpreted as prolactinoma36,38,42. Concomi
tant hyperprolactinemia and lactotroph hyperplasia is common in severe thyrotroph hyperplasia1,2,38,40. A decline in the number of somatotrophs is seen in cases of massive thyrotroph hyperplasia1. Both the transformation of thyrotroph hyperplasia into and its coexistence with thyrotroph adenoma have been reported2.
CLINICAL SIGNIFICANCE
The incidence of pituitary hyperplasia in general and the frequency of its subtypes are not known. This primarily relates to the difficulties in establishing the diagnosis as outlined above. One study with a limited number of patients found lactotroph hyperplasia to be present in about 20%, somatotroph hyperplasia in 13%, corticotroph hyperplasia in 13%, and thyrotroph hyperplasia in 7% of all normal pituitaries56. In a study of two hundred fifty autopsy pituitaries, PI derived POMC cells hyperplasia was found to be present in 29% of men and 14% of women54. Many pituitary hyperplasias might be clinically silent. Focal pituitary hyperplasia is usually an incidental finding in autopsy, has no apparent clinical counterpart and is of no clinical significance1. Some pituitary cell type hyperplasias, such as PI derived POMC-cells hyperplasia, mostly constitute an incidental finding that is common in old age1,54. Similarly, gonadotroph hyperplasia is not usually associated with the primary cause of pituitary disease and is thought to be of no clinical significance, though rare cases of hypergonadotropic ovarian failure secondary to gonadotroph hyperplasia have been reported44,45. As outlined above, pituitary hyperplasia could be a physiologic response to external stimuli that are appropriate and warranted. Physiologic pituitary enlargement as a result of hyperplasia, such as those seen with pregnancy, is something that needs to be borne in mind when interpreting the magnetic resonance imaging of the sella in patients undergoing those physiologic states. Failure to do so may result in erroneous diagnoses. In some circumstances, the response of pituitary cells to ongoing stimuli can turn into a clinically significant problem. This is clearly demonstrated by massive thyrotroph hyperplasia with intracranial hypertension, compression of the adjacent structures and hormonal disturbance as a response to prolonged primary hypothyroidism. It is important to realize that pituitary hyperplasia can be large enough to mimic pituitary adenoma, since the first can be managed medically most of the time thus avoiding unnecessary surgery. Pituitary cell hyperplasia can be a source of hyperprolactinemia, gigantism and Cushing's disease. Indeed, up to 18% of Cushing's disease and some of the nonremissions following successful removal of corticotroph adenoma could be attributed to corticotroph hyperplasia56.
Pituitary hyperplasia can occur in coexistence with pituitary adenoma. This association between hyperplasia and neoplasia has been observed for somatotroph, lactotroph, thyrotroph and POMC-cell, but even more often in the case of corticotroph cell hyperplasia1,2,54,56,60,66. Neoplastic transformation from pituitary hyperplasia is rare though well documented1,2,21,28,66. The question of the co-existence versus progression from hyperplasia to neoplasia is difficult to answer. The presence of tumorlets, small monomorphous proliferations, associated with focal dissolution of normal tissue architecture with the appearance of microadenoma but with distended and yet preserved acini in several hyperplastic lesions is strongly suggestive of the progression theory1,28. Evidence exists indicating that persistent hyperstimulation (e.g due to overproduction of releasing hormones or loss of dopaminergic inhibition) may lead the hyperplastic cell to acquire autonomy and progress to neoplasm1,21,28,38,61. Though pituitary hyperplasia is not a prerequisite for the formation of pituitary adenoma, the hyperplastic cells are more susceptible to neoplastic transformation than normal cells.
DIFFERENTIAL DIAGNOSIS
The main two differentials of pituitary hyperplasia are normal gland and pituitary adenoma. The diffuse morphologic type is the one that is more likely to be confused with normal gland while the nodular type can mimic pituitary adenoma. To reach the correct diagnosis in the first scenario, it is important to correlate the clinical, radiological and pathological findings. The hallmark of diffuse pituitary hyperplasia is increased cell number of mostly one subtype of pituitary cells accompanied by variable enlargement of acini. The features that distinguish nodular hyperplasia from adenoma have been discussed above and include, in the case of hyperplasia polyclonality, preservation of acini, lack of clear demarcation and lack of compressive effect.
PROGNOSIS AND MANAGEMENT
The prognosis of pituitary hyperplasia depends on its cause, endocrine effect and extent. Most of the time it is reversible by eliminating the underlying cause. This leaves only a minor role for surgery as a diagnostic tool or for treatment of exceptional case scenarios such as thyrotroph hyperplasia with severe symptoms from mass effect not responding to thyroxine replacement or rare cases of Cushing's disease with severe endocrine imbalance. It is clear that in cases of pituitary hyperplasia secondary to target organs failure, the earlier the medical management, in the form of replacement therapy, the better is the prognosis.
CONCLUSION
Pituitary hyperplasia, though rare, is an important condition. Understanding the various subtypes, morphologic forms and pathologic features help to make the diagnosis. Despite the diagnostic difficulties, reaching the correct diagnosis is of vital importance in this condition which is usually medically treatable, thus avoiding unnecessary surgery. Understanding pituitary hyperplasia is also an important link to understanding the tumorigenesis in the pituitary gland.
REFERENCES
1. Horvath E, Kovacs K, Scheithauer BW, 1999 Pituitary hyperplasia. Pituitary 1: 169-179.
2. Scheithauer BW, Horvath E, Lloyd RV, Kovacs K, 1998 Pituitary Hyperplasia. Pathol Case Rev 3: 281-289.
3. Horvath E, 1988 Pituitary hyperplasia. Pathol Res Pract 183: 623-625.
4. McNicol AM, Carbajo-Perez E, 1999 Aspects of anterior pituitary growth, with special reference to corticotrophs. Pituitary 1: 257-268.
5. Khalil A, Kovacs K, Sima AA, Burrow GN, Horvath E, 1984 Pituitary thyrotroph hyperplasia mimicking prolactin-secreting adenoma. J Endocrinol Invest 7: 399-404.
6. Adams C, Dean HJ, Israels SJ, Patton A, Fewer DH, 1994 Primary hypothyroidism with intracranial hypertension and pituitary hyperplasia. Pediatr Neurol 10: 166-168.
7. Alkhani AM, Cusimano M, Kovacs K, Bilbao JM, Horvath E, Singer W, 1999 Cytology of pituitary thyrotroph hyperplasia in protracted primary hypothyroidism. Pituitary 1: 291-295.
8. Dadachanji MC, Bharucha NE, Jhankaria BG, 1994 Pituitary hyperplasia mimicking pituitary tumor. Surg Neurol 42: 397-399.
9. Preisig P, 1999 What makes cells grow larger and how do they do it? Renal hypertrophy revisited. Exp Nephrol 7: 273-283.
10. Levy L, Alvaro V, Dubray C, Joubert D, 1994 Ca(2+)-dependent protein kinase C isoforms in rat pituitary hyperplasia: effect of in vivo treatment with quinagolide. Eur J Pharmacol 268: 327-334.
11. Lloyd RV, Ruebel KH, Zhang S, Jin L, 2002 Pituitary hyperplasia in glycoprotein hormone alpha subunit-, p18(INK4C)-, and p27(kip-1)-null mice: analysis of proteins influencing p27(kip-1) ubiquitin degradation. Am J Pathol 160: 1171-1179.
12. Qian X, Jin L, Grande JP, Lloyd RV, 1996 Transforming growth factor-beta and p27 expression in pituitary cells. Endocrinology 137: 3051-3060.
13. Foster JR, 2000 Cell death and cell proliferation in the control of normal and neoplastic tissue growth. Toxicol Pathol 28: 441-446.
14. Levy A, 1999 Mitosis and apoptosis in the pituitary gland: tumour formation or hyperplasia? Baillieres Best Pract Res Clin Endocrinol Metab 13: 353-365.
15. Levy A, 2002 Physiological implications of pituitary trophic activity. J Endocrinol 174: 147-155.
16. Cameron HA, McEwen BS, Gould E, 1995 Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. J Neurosci 15: 4687-4692.
17. Gonzalez S, Mauriello-Romanazzi G, Berrendero F, Ramos JA, Franzoni MF, Fernandez-Ruiz J, 2000 Decreased cannabinoid CB1 receptor mRNA levels and immunoreactivity in pituitary hyperplasia induced by prolonged exposure to estrogens. Pituitary 3: 221-226.
18. Jin L, Burguera BG, Couce ME, et al, 1999 Leptin and leptin receptor expression in normal and neoplastic human pituitary: evidence of a regulatory role for leptin on pituitary cell proliferation. J Clin Endocrinol Metab 84: 2903-2911.
19. Lloyd RV, Jin L, Tsumanuma I, et al, 2001 Leptin and leptin receptor in anterior pituitary function. Pituitary 4: 33-47.
20. Nedvidkova J, Pacak K, Haluzik M, Nedvidek J, Schreiber V, 2001 The role of dopamine in methylene blue-mediated inhibition of estradiol benzoate-induced anterior pituitary hyperplasia in rats. Neurosci Lett 304: 194-198.
21. Saiardi A, Bozzi Y, Baik JH, Borrelli E, 1997 Antiproliferative role of dopamine: loss of D2 receptors causes hormonal dysfunction and pituitary hyperplasia. Neuron 19: 115-126.
22. Ezzat S, 2001 The role of hormones, growth factors and their receptors in pituitary tumorigenesis. Brain Pathol 11: 356-370.
23. Asa SL, Ramyar L, Murphy PR, Li AW, Ezzat S, 2001 The endogenous fibroblast growth factor-2 antisense gene product regulates pituitary cell growth and hormone production. Mol Endocrinol 15: 589-599.
24. Borrelli E, Sawchenko PE, Evans RM, 1992 Pituitary hyperplasia induced by ectopic expression of nerve growth factor. Proc Natl Acad Sci U S A 89: 2764-2768.
25. Cameron HA, Hazel TG, McKay RD, 1998 Regulation
of neurogenesis by growth factors and neurotransmitters. J Neurobiol 36: 287-306.
26. Gonzalez IA, Diaz-Torga G, Piroli G, et al, 2000 Bromocriptine restores angiotensin II response in pituitary hyperplasia. Mol Cell Endocrinol 165: 67-74.
27. Cameron HA, Gould E, 1994 Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 61: 203-209.
28. Zimmerman D, Young WF Jr, Ebersold MJ, et al, 1993 Congenital gigantism due to growth hormone-releasing hormone excess and pituitary hyperplasia with adenomatous transformation. J Clin Endocrinol Metab 76: 216-222.
29. Asa SL, Scheithauer BW, Bilbao JM, et al, 1984 A case for hypothalamic acromegaly: a clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J Clin Endocrinol Metab 58: 796-803.
30. Mayo KE, Hammer RE, Swanson LW, Brinster RL, Rosenfeld MG, Evans RM, 1988 Dramatic pituitary hyperplasia in transgenic mice expressing a human growth hormone-releasing factor gene. Mol Endocrinol 2: 606-612.
31. Burton FH, Hasel KW, Bloom FE, Sutcliffe JG, 1991 Pituitary hyperplasia and gigantism in mice caused by a cholera toxin transgene. Nature 350: 74-77.
32. Asa SL, Kovacs K, Tindall GT, Barrow DL, Horvath E, Vecsei P, 1984 Cushing's disease associated with an intrasellar gangliocytoma producing corticotrophin-releasing factor. Ann Intern Med 101: 789-793.
33. Clayton R, Burden AC, Schrieber V, Rosenthal FD, 1977 Secondary pituitary hyperplasia in Addison's disease. Lancet 2: 954-956.
34. Floyd JL, Dorwart RH, Nelson MJ, Mueller GL, DeVroede M, 1984 Pituitary hyperplasia secondary to thyroid failure: CT appearance. AJNR Am J Neuroradiol 5: 469-471.
35. Hutchins WW, Crues JV III, Miya P, Pojunas KW, 1990 MR demonstration of pituitary hyperplasia and regression after therapy for hypothyroidism. AJNR Am J Neuroradiol 11: 410.
36. Khalil A, Kovacs K, Sima AA, Burrow GN, Horvath E, 1984 Pituitary thyrotroph hyperplasia mimicking prolactin-secreting adenoma. J Endocrinol Invest 7: 399-404.
37. Okuno T, Sudo M, Momoi T, et al, 1980 Pituitary hyperplasia due to hypothyroidism. J Comput Assist Tomogr 4: 600-602.
38. Pioro EP, Scheithauer BW, Laws ER Jr, Randall RV, Kovacs KT, Horvath E, 1988 Combined thyrotroph and lactotroph cell hyperplasia simulating prolactin-secreting pituitary adenoma in long-standing primary hypothyroidism. Surg Neurol 29: 218-226.
39. Shingyouchi H, Shindo M, Kobayashi M, et al, 1990 [Pituitary hyperplasia in primary hypothyroidism]. Rinsho Hoshasen 35: 529-532.
40. Williams RS, Williams JP, Davis MR, Hutto RL, 1990 Primary hypothyroidism with pituitary hyperplasia and basal ganglia calcifications. Clin Imaging 14: 330-332.
41. Yamagishi S, Yokoyama-Ohta M, 1999 A rare case of pituitary hyperplasia with suprasellar extension due to primary myxoedema: case report. J Int Med Res 27: 49-52.
42. Yamamoto Y, Kunishio K, Sunami N, Yamamoto Y, Asari S, Yunoki S, 1987 [A case of pituitary hyperplasia associated with primary hypothyroidism]. No Shinkei Geka 15: 903-908. 43. Young M, Kattner K, Gupta K, 1999 Pituitary hyperplasia resulting from primary hypothyroidism mimicking macroadenomas. Br J Neurosurg 13: 138-142.
44. Okuda K, Yoshikawa M, Sugiyama S, et al, 1987 [Hypergonadotropic ovarian failure in three patients with pituitary hyperplasia]. Nippon Sanka Fujinka Gakkai Zasshi 39: 1579-1584.
45. Okuda K, Yoshikawa M, Ushiroyama T, Sugimoto O, Maeda T, Mori H, 1989 Two patients with hypergonadotropic ovarian failure due to pituitary hyperplasia. Obstet Gynecol 74: 498-501.
46. Radner H, Pummer K, Lax S, Wandschneider G, Hofler H, 1991 Pituitary hyperplasia after goserelin (LHRH-analogue) therapy. Neuropathol Appl Neurobiol 17: 75-81.
47. Riedl S, Frisch H, 1997 Pituitary hyperplasia in a girl with gonadal dysgenesis and primary hypothyroidism. Horm Res 47: 126-130.
48. Kido G, Miyagi A, Shibuya T, Miyagami M, Tsubokawa T, Sawada T, 1994 [Turner's syndrome with pituitary hyperplasia: a case report]. No Shinkei Geka 22: 333-338.
49. Lloyd RV, Schmidt K, Nath V, 1985 Effects of pergolide on diethylstilbestrol-induced rat pituitary hyperplasia. Am J Pathol 121: 486-495.
50. Maeda T, Sawada K, Itoh Y, Moriwaki K, Mori H, 1991 Decreased prolactin level in secretory granules and their increased exocytosis in estrogen-induced pituitary hyperplasia in rats treated with a dopamine agonist. Lab Invest 65: 679-687.
51. Terry LC, Craig R, Hughes T, et al, 1985 Hypothalamic monoaminergic activity and pituitary function in male rats with estrogen-induced pituitary hyperplasia. Neuroendocrinology 41: 269-275.
52. Suarez C, Garcia Tornadu I, Khalil W, Becu-Villalobos D, 2002 Dehydroepiandrosterone treatment attenuates oestrogen-induced pituitary hyperplasia. J Endocrinol 174: 447-454.
53. Duskova J, Povysil C, Schreiber V, 1997 AgNORs in estrogen-induced and lisuride- and thyreoidin-inhibited rat anterior pituitary hyperplasia. Computer image analysis and nonautomated AgNOR typing. Anal Quant Cytol Histol 19: 55-61.
54. Horvath E, Kovacs K, Lloyd RV, 1999 Pars intermedia of the human pituitary revisited: morphologic aspects and frequency of hyperplasia of POMC-peptide immunoreactive cells. Endocr Pathol 10: 55-64.
55. Asa SL, Penz G, Kovacs K, Ezrin C, 1982 Prolactin cells in the human pituitary. A quantitative immunocytochemical analysis. Arch Pathol Lab Med 106: 360-363.
56. Saeger W, Ludecke DK, 1983 Pituitary hyperplasia. Definition, light and electron microscopical structures and significance in surgical specimens. Virchows Arch A Pathol Anat Histopathol 399: 277-287.
57. Vidal S, Horvath E, Kovacs K, Cohen SM, Lloyd RV, Scheithauer BW, 2000 Transdifferentiation of somatotrophs to thyrotrophs in the pituitary of patients with protracted primary hypothyroidism. Virchows Arch 436: 43-51.
58. Vidal S, Horvath E, Kovacs K, Lloyd RV, Smyth HS, 2001 Reversible transdifferentiation: interconversion of somatotrophs and lactotrophs in pituitary hyperplasia. Mod Pathol 14: 20-28.
59. Bloodworth JM Jr, 1988 Assessment of the pituitary hyperplasia/neoplasia interface. Pathol Res Pract 183: 626-630.
60. Peillon F, Dupuy M, Li JY, et al, 1991 Pituitary enlargement with suprasellar extension in functional hyperprolactinemia due to lactotroph hyperplasia: a pseudotumoral disease. J Clin Endocrinol Metab 73: 1008-1015.
61. Brinkmeier ML, Stahl JH, Gordon DF, et al, 2001 Thyroid hormone-responsive pituitary hyperplasia independent of somatostatin receptor 2. Mol Endocrinol 15: 2129-2136.
62. Papakonstantinou O, Bitsori M, Mamoulakis D, Bakantaki A, Papadaki E, Gourtsoyiannis N, 2000 MR imaging of pituitary hyperplasia in a child with growth arrest and primary hypothyroidism. Eur Radiol 10: 516-518.
63. Vaidya RA, Aloorkar SD, Raikar RS, et al, 1977 Functional pituitary hyperplasia in primary hypothyroidism: normalisation of visual field defects on thyroid replacement in a girl with galactorrhoea—amenorrhoea. J Assoc Physicians India 25: 923-928.
64. Wolansky LJ, Leavitt GD, Elias BJ, Lee HJ, Dasmahapatra A, Byrne W, 1996 MRI of pituitary hyperplasia in hypothyroidism. Neuroradiology 38: 50-52.
65. Kovacs K, Horvath E, 2001 The differential diagnosis of lesions involving the sella turcica. Endocr Pathol 12: 389-395.
66. Ferramosca B, Bertoni F, Ravaglia G, Lodi A, 1976 [Adenomatous transformation of acidophilic pituitary hyperplasia in acromegaly]. Minerva Med 67: 3011-3020.
Address correspondence and requests for reprints to:
Mubarak Al-Gahtany, M.D., 1505-280 Simcoe Street.,
Toronto, ON, Canada M5T 2Y5, Fax: +1 416 977-4773,
e-mail: mubarak.al.gahtany@utoronto.ca
Received 10-04-03, Revised 27-05-03, Accepted 06-06-03