1Section on Endocrinology and Genetics, NICHD, National Institutes of Health, Bethesda, MD, USA; 2Pediatric endocrinology
training program, NICHD, National Institutes of Health, Bethesda, MD, USA; 3Department of Pediatrics, University of
Tennessee Health Science Center, Memphis, TN, USA
*SU and YHJ equally contributed to this
manuscript.
BACKGROUND: Tetrasomy X is a rare chromosomal aneuploidy seen in girls,
associated with facial dysmorphism, premature ovarian insufficiency and
intellectual disability. A Rathke’s cleft cyst (RCC) is a remnant of Rathke’s
pouch which may cause multiple pituitary hormone deficiencies by exerting
pressure on the pituitary gland in the sella.
METHODS/RESULTS:
The patient was diagnosed with tetrasomy X by karyotyping during infancy. Brain
MRI and multiple endocrine stimulation tests revealed RCC and combined
pituitary hormone deficiency (growth hormone deficiency, secondary
adrenal insufficiency and central hypothyroidism) likely due to RCC.
CONCLUSION: We report the first case in the
literature of a girl with 48, XXXX and combined pituitary hormone deficiency
due to Rathke’s cyst.
Combined pituitary hormone deficiency, Κaryotype, Rathke’s cleft cyst, SNP array, Tetrasomy X
INTRODUCTION
Tetrasomy X is a rare chromosomal aneuploidy seen in girls which was first described in 1961 by Carr et al,1 since which time only a little over 100 cases have been reported.2 The clinical spectrum of tetrasomy includes facial dysmorphism, premature ovarian insufficiency and intellectual disability, and, according to case reports, skeletal abnormalities, aplastic anemia and systemic lupus erythematous.3-4 Some characteristic facial features seen are epicanthic folds, flat nasal bridge, hypertelorism and nystagmus. Mental retardation is characteristic of 48, XXXX, the average IQ being 60.3 Individuals have been described as pleasant, friendly and cooperative but also socially inappropriate, aggressive and emotionally labile, which may continue until adulthood.3
A Rathke’s cleft cyst (RCC) is a remnant of Rathke’s pouch which is derived from ectoderm and extends upward to fuse with the infundibulum that moves downward from neuroectoderm during early gestation.5 RCC is usually to be found between the anterior and posterior pituitary gland either in the sellar or suprasellar region.5 Although several hypotheses of the derivation of RCC have been proposed, the most widely accepted mechanism is failed regression of the cleft cyst.6 RCCs are mostly a non-neoplastic lesion lined with a single layer of cuboidal epithelium containing mucoid material and are often discovered incidentally.7 We hereby report a unique case in the literature of a girl with 48, XXXX and RCC who developed early growth hormone deficiency and later developed partial secondary adrenal insufficiency and central hypothyroidism.
CASE PRESENTATION
The patient was born full term at 40 weeks via normal spontaneous vaginal delivery to a 32-year old mother and 33-year old father. There were no perinatal complications and the Apgar scores were 4 and 9 at 5 and 10 minutes of life, respectively. She was small for gestational age with a birth weight of 5 lbs. 2 oz. at the 5th percentile and birth length of 18.5 inches between the 5th and 10th percentile. The patient was noted as having dysmorphic features including hypertelorism, low-set ears, epicanthal folds and a flat nasal bridge. During her first year of life she was diagnosed with failure to thrive, including a progressive deceleration in growth rate and global developmental delay. At 14 months of age, her facial dysmorphism, poor growth and developmental delay prompted additional evaluation including a karyotype that revealed tetrasomy X (Figure 1A). The parents’ karyotypes were normal. A SNP array was also performed to evaluate the characteristics of the extra X chromosomes (Figure 1B). Her laboratory evaluation was remarkable for a low IGF-1 of 37ng/mL (reference range 49-342 ng/mL) and her bone age was delayed (1 month at chronological age of 17 months (Figure 2A). She also underwent a brain and pituitary MRI which showed delayed demyelination and an 8 X 4 X 13 mm cystic lesion in the sella turcica, indicating a Rathke’s cleft cyst (Figure 2B).
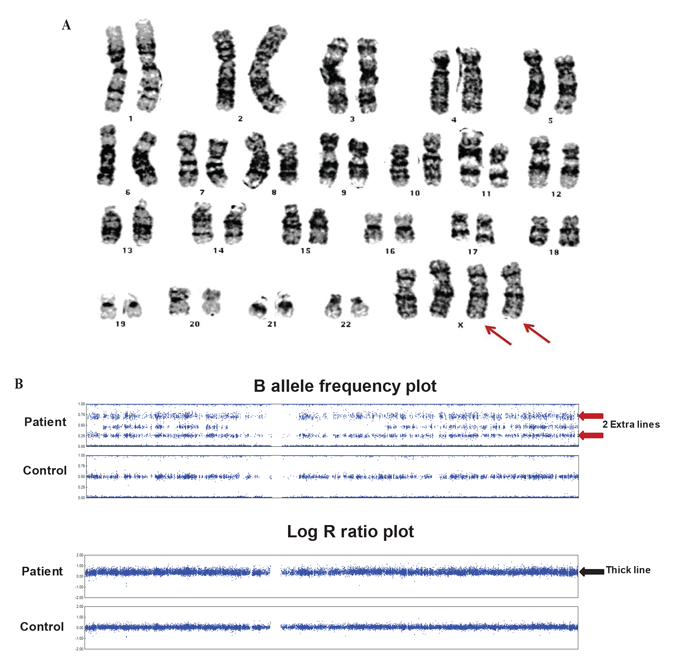
Figure 1. Karyotype and SNP array. A) Karyotype showed 2 extra X chromosomes (red arrows). B) X chromosomes on SNP array shows 2 extra lines on B allele frequency plot (red arrows) compared to control showing heterozygous SNPs are separated into 3 distinct lines. The corresponding log R ratio plot shows a thicker line with higher log R ratio (black arrow) than control because extra chromosomes are calculated as duplication.

Figure 2. Bone age and MRI images of Rathke’s cleft cyst. A) Bone age x-ray at chronological age of 17 months. A T1 sagittal scan is represented showing a non-enhancing 8 × 4 × 14 mm cystic lesion in the pituitary gland (yellow arrow). B) 17 months old. C) 7 years 10 months.
With progressively declining growth rate, a low IGF-1 and a Rathke’s cleft cyst in the pituitary gland, growth hormone deficiency was strongly suspected, although this could not be confirmed via the growth hormone stimulation test due to difficulties in securing vascular access. Therefore, under clinical suspicion, she was diagnosed with growth hormone deficiency and at 21 months of age she started treatment requiring a low dose of growth hormone (0.13 mg/kg/week), to which she responded with improvement in IGF-1 and growth velocity, as is typical in this management of growth hormone deficiency (Figure 3A). At the time, her thyroid function tests and cortisol peak of 35.1 ug/dL after the standard 250 mcg ACTH stimulation test were appropriately within normal range. Serial MRIs performed over the years showed no change in the size of the cyst (Figure 2C).
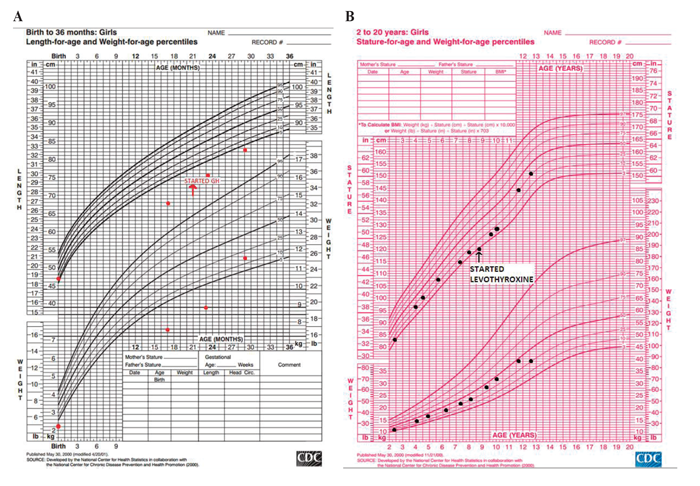
Figure 3. Growth charts of patient. A) Infantile growth chart (0-2 years). B) Childhood growth chart (2-20 years).
At her 8 years and 8 months follow-up visit, she again presented with height deceleration. On further evaluation, diurnal TSH showed a peak increment TSH of 41%, suggesting central hypothyroidism (normal increment >48%).8 Her ACTH stimulation test showed a peak cortisol of 13.9 mcg/dL at 60 minutes, indicating partial adrenal insufficiency. A physiologic dose of hydrocortisone and of levothyroxine replacement were initiated and her growth improved accordingly (Figure 3B). At age 8, she was prescribed aripiprazole for behavioral outbursts and subsequently had significant weight gain, her BMI percentile increasing from 50th to 85th. The patient had minimal breast development in addition to lipomastia at age 8 years, which was thought to be secondary to peripheral conversion of estrogen from extra adipose tissue: her breasts gradually regressed with dietary control. Her estradiol level was below detection limits. She restarted breast development at age 10 and reached Tanner 3 at age 11 years and 9 months. At that time, she developed an episode of vaginal bleeding which lasted 5 days but did not recur. Her LH was 5.9 U/L, FSH was 2.6 U/L and estradiol was 31.6 pg/mL (Table 1).

Figure 4. Patient features. Pictures were taken at 15 months (A) and 12 years 8 months (B). Dysmorphic features including hypertelorism, epicanthal folds and a flat nasal bridge are shown.
At the age of 12 years 7 months, she had yet to have a second menses, despite having FSH 7.5 U/L and LH 3.8 U/L appropriate for her age and Tanner stage 4 breast development, though with inappropriately low estradiol of <5.0 pg/mL. At the age of 13 years 5 months, she had had a total of 3 menses and her estradiol was normal for her Tanner stage (Table 1). Her bone age was 12 years 9 months at a chronological age of 12 years 7 months old, showing a predicted adult height of 157 cm, which is close to her mid-parental height of 156 cm. The patient has had no evidence of posterior pituitary dysfunction and no biochemical evidence of diabetes insipidus. Her other medical problems included chronic constipation, enuresis and sleep apnea, for which a BiPAP machine is being used. Her intelligence quotient was 60 at 8 years of age. The patient also has had tic-like movements, such as eye blinking or biting lips which have intensified with age. Her behavioral and psychological disorders after onset of puberty have been challenging to control and are associated with episodes of unprovoked yelling and crying. Since she showed self-injurious behavior, a psychiatry consultation was performed for behavioral intervention. The patient is currently in the sixth grade at a school with self-contained classes. The patient’s photograph is shown in Figure 4.
DISCUSSION
Our patient has a 48, XXXX karyotype and combined pituitary hormone deficiency due to a RCC. While tetrasomy X itself is a rare condition, no case associated with symptomatic RCC has been reported in the medical literature, making this patient a unique case. The patient showed typical characteristics of tetrasomy X: facial dysmorphism, intellectual disability and likely premature ovarian insufficiency. As is well known, one of the interesting effects of having extra chromosomes is the height outcome. It is postulated that patients with extra sex chromosomes are tall, since X and Y chromosomes carry the short stature homeobox gene of the X chromosome (SHOX) in the Pseudo Autosomal Region (PAR), which is reflected in its positive dose-dependent effect on height. For example, children with Turner syndrome (loss of X chromosome) often present with short stature, while children with Klinefelter syndrome (extra X syndrome) present with tall stature. However, Ottesen et al recently found a non-linear effect of the number of sex chromosomes on height, which is that in the presence of 4 or more sex chromosomes a decrease in the height of patients is seen, as opposed to the increasing heights that are expected.9 It is speculated that the presence of a duplication of this genomic region does not always result in tall stature because the height outcome may be affected by the size of duplication or position of the duplication, including flanking sequences or involvement of transcriptional enhancers.9 In patients with tetrasomy X, there seems to be variability in height as they can show normal growth, short stature or tall stature.9 Our patient presented with failure to thrive and poor growth during early infancy, which required growth hormone treatment, probably due to growth hormone deficiency. On growth hormone, her height has been between the 25th to 50th percentiles for age. Pubertal development in our patient was also unique. She has delayed puberty, having FSH and LH levels within normal range but undetectable estradiol levels, which eventually recovered showing appropriate FSH, LH and estradiol levels for her Tanner stage. It has been reported that with an increasing number of X chromosomes, the frequency of premature ovarian insufficiency increases, and the age at presentation decreases, possibly due to earlier depletion of ovarian follicles.10 Therefore, it has been suggested that patients with tetrasomy X should be screened for ovarian insufficiency during early adolescence, as hormone replacement therapy may be required for the development of secondary sex characteristics and for prevention of eventual osteoporosis.10 However, some patients with 48, XXXX may have normal ovarian function.11
RCC has rarely been reported in children and adolescents12 and may not be discovered clinically until late adulthood.13 RCC is commonly found in 11-22% of autopsies and mostly remains asymptomatic.14 In about 30% of patients, RCC may increase in size requiring close monitoring with pituitary MRI.15 When symptomatic, the common clinical symptoms in pediatric cases are headaches, visual disturbance and endocrine dysfunction.16-17 Although these symptoms are very rare in children with RCC, if they develop endocrine dysfunction, these patients could present with growth hormone deficiency, hyperprolactinemia, central precocious puberty, diabetes insipidus, secondary adrenal insufficiency, secondary amenorrhea or central hypothyroidism.16-23 45-60% of children with symptomatic RCC may develop endocrine abnormalities,22 but not all endocrine abnormalities would resolve after surgical removal of RCC.24 It is of interest that our patient developed growth hormone deficiency due to RCC at a very early age but the rest of her pituitary hormone deficiencies during childhood. It has been hypothesized that pituitary defects in patients with RCC are mainly related to chronic compression of the gland by the cyst filled with mucoid material.5 It could therefore be speculated that the mass effect might occur as early as infancy or even in early intrauterine life, as seen in a case with micropenis and growth hormone deficiency secondary to RCC.25 We have ascertained from our thorough literature review that this is the first recorded case of a girl with 48, XXXX and symptomatic RCC and we conclude that the patient’s RCC is most probably an unrelated event, independent of the tetrasomy X.
REFERENCES
1. Carr DH, Barr ML, Plunkett ER, 1961 An XXXX sex chromosome complex in two mentally defective females. Can Med Assoc J 84: 131-137.
2. Samango-Sprouse C, Keen C, Mitchell F, Sadeghin T, Gropman A, 2015 Neurodevelopmental variability in three young girls with a rare chromosomal disorder, 48, XXXX. Am J Med Genet Part A 167A: 2251-2259.
3. Linden MG, Bender BG, Robinson A, 1995 Sex chromosome Tetrasomy and Pentasomy. Pediatrics 96: 672-682.
4. Alvaro-Gracia JM, Humbria A, García-Vicuña R, Ariza A, García-Vadillo A, Laffón A, 1989 Systemic lupus erythematosus and tetrasomy-X. J Rheumatol 16: 1486-1488.
5. Han SJ, Rolston JD, Jahangiri A, Aghi MK, 2014 Rathke’s cleft cysts review of natural history and surgical outcomes. J Neurooncol 117: 197-203.
6. Mendelson ZS, Husain Q2, Kanumuri VV, Eloy JA, Liu JK, 2015 Endoscopic transsphenoidal surgery of Rathke’s cleft cyst. J Clin Neurosci 22: 149-154.
7. Trifanescu R, Ansorge O, Wass JA, Grossman AB, Karavitaki N, 2012 Rathke’s cleft cysts Clin Endocrinol (Oxf) 76: 151-160.
8. Rose SR, Manasco PK, Pearce S, Nisula BC, 1990 Hypothyroidism and deficiency of the nocturnal thyrotropin surge in children with hypothalamic-pituitary disorders. J Clin Endocrinol Metab 70: 1750-1755.
9. Ottesen AM, Aksglaede L, Garn I, et al, 2010 Increased number of sex chromosomes affects height in a nonlinear fashion: A study of 305 patients with sex chromosome aneuploidy. Am J Med Genet Part A 152A: 1206-1212.
10. Kara C, Üstyol A, Yılmaz A, Altundağ E, Oğur G, 2014 Premature ovarian failure due to Tetrasomy X in an adolescent girl. Eur J Pediatr 173: 1627-1630.
11. Rooman RP, Van Driessche K, Du Caju MV, 2002 Growth and ovarian function in girls with 48, XXXX karyotype--patient report and review of the literature. J Pediatr Endocrinol Metab 15: 1051-1055.
12. Jahangiri A, Molinaro AM, Tarapore PE, et al, 2012 Rathke cleft cysts in pediatric patients: presentation, surgical management, and postoperative outcomes. Neurosurg Focus 31: E3.
13. Isono M, Kamida T, Kobayashi H, Shimomura T, Matsuyama J, 2001 Clinical features of symptomatic Rathke’s cleft cyst. Clin Neurol Neurosurg 103: 96-100.
14. Teramoto A, Hirakawa K, Sanno N, Osamura Y, 1994 Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology 193: 161-164.
15. Culver SA, Grober Y, Ornan DA, et al, 2015 A Case for Conservative Management: Characterizing the Natural History of Radiographically Diagnosed Rathke Cleft Cysts. J Clin Endocrinol Metab 100: 3943-3948.
16. Bizzarri C, Marini R, Ubertini G, Cappa M, 2012 Partially Reversible Hypopituitarism in an Adolescent with a Rathke Cleft Cyst. Clin Pediatr Endocrinol 21: 75-80.
17. Lim HH, Yang SW, 2010 Risk factor for pituitary dysfunction in children and adolescents with Rathke’s cleft cysts. Korean J Pediatr 53: 759-765.
18. Oh YJ, Park HK, Yang S, Song JH, Hwang IT, 2014 Clinical and radiological findings of incidental Rathke’s cleft cysts in children and adolescents. Ann Pediatr Endocrinol Metab 19: 20-26.
19. Zada G, Ditty B, McNatt SA, McComb JG, Krieger MD, 2009 Surgical treatment of Rathke cleft cysts in children. Neurosurgery 64: 1132-1137.
20. Evliyaoglu O, Evliyaoglu C, Ayva S, 2010 Rathke cleft cyst in seven-year-old girl presenting with central diabetes insipidus and review of literature. J Pediatr Endocrinol Metab 23: 525-529.
21. Iannelli A, Martini C, Cosottini M, Castagna M, Bogazzi F, Muscatello L, 2012 Rathke’s cleft cysts in children: clinical, diagnostic, and surgical features. Childs Nerv Syst 28: 297-303.
22. Briceño LG, Gunczler P, Solis O, 2012 Rathke Cleft Cyst as Cause of Growth Hormone Deficiency in a 9-Year-Old Girl J Pediatr 160: 708-708.e1
23. Katavetin P, Cheunsuchon P, Grant E, et al, 2010 Rathke’s cleft cysts in children and adolescents: association with female puberty. J Pediatr Endocrinol Metab 23: 1175-1180.
24. Lim HH, Yang SW, 2010 Risk factor for pituitary dysfunction in children and adolescents with Rathke’s cleft cysts. Korean J Pediatr 53: 759-765.
25. Setian N, Aguiar CH, Galvão JA, Crivellaro CE, Dichtchekenian V, Damiani D, 1999 Rathke’s cleft cyst as a cause of growth hormone deficiency and micropenis. Child’s Nerv Syst 15: 271-273.
Address for correspondence:
Constantine A. Stratakis, MD, D(med)Sci, Chief, Section on Genetics & Endocrinology (SEGEN), Program on Developmental Endocrinology & Genetics (PDEGEN), NICHD, NIH, Building 10, CRC, East Laboratories, Rm. 1-3330 (1-3216 lab), 10 Center Drive, Bethesda, MD 20892, USA; Tel.: 301-4964686 (sec)/301-4021998 (lab), Fax: 301-4020574, E-mail: stratakc@mail.nih.gov
Received: 17-02-2017, Accepted: 02-03-2017