1Department of Endocrinology, Diabetes and Metabolism, 2Genetics Department; Faculty of Medicine University of Porto, Centro Hospitalar São João, Porto, Portugal
Adipogenesis, Genetic or acquired diseases, Insulin resistance, Lipodystrophy, PPARG
Sir,
Familial partial lipodystrophy is an autosomal dominant genetic disorder characterized by lipoatrophy of the extremities and gluteal region and lipohypertrophy of the face, neck and/or trunk. It is associated with insulin resistance, hypertriglyceridemia and increased risk of recurrent episodes of pancreatitis. The PPARG mutation forms, called familial partial lipodystrophies type 3, are very rare, accounting for only approximately 30 patients. We report here the case of a 60-year old Caucasian woman who had started to develop gradual fat loss of the extremities and gluteal region and facial and abdominal fat increase at 30 years of age. Subsequently, type V dyslipidemia and diabetes were diagnosed and she developed acute pancreatitis. Physical examination confirmed clinical lipodystrophy. A genetic study revealed a heterozygous mutation in the PPARG gene (p.Gly161Val; c.482G>T), not previously described. In conclusion, clinical appearance and past medical history in conjunction with the genetic study led to the identification of a novel gene mutation in PPARG, enabling the diagnosis of familial partial lipodystrophy type 3. This case highlights the importance of early recognition of a lipodystrophy syndrome in order to prevent metabolic complications, recurrent pancreatitis and the onset of cardiovascular disease.
Lipodystrophies are rare syndromes characterized by abnormal fat tissue deposition.1,2 The hallmark of these disorders is a selective loss of subcutaneous and visceral adipose tissue. Lypodystrophies can be categorized into different types: partial or complete and acquired or genetic, which explain the considerable heterogeneity of fat mass distribution and degree of fat loss between lipodystrophy forms. Despite these different phenotypes, lipodystrophies present a common spectrum of metabolic disorders, such as insulin resistance, diabetes mellitus, hypertriglyceridemia, hepatic steatosis, ovary polycystic syndrome and acanthosis nigricans. The severity of metabolic complications is correlated with the extent of adipose tissue loss.3
Familial partial lipodystrophies (FPLD) are autosomal dominant inherited forms that might be due to different mutations. The first gene identified was LMNA on chromosome 1q21–22.4 Lamins A and C are nuclear lamina proteins whose mutations result in the premature death of adipocytes, being responsible for the Dunnigan variety which appears to represent the commonest form.5 Subsequently, four other candidate genes were identified: peroxisome proliferator-activated receptor gamma (PPARG),6 v-AKT murine thymoma oncogene homolog 2 (AKT2),7 regulator of insulin signaling, cell death-inducing DNA fragmentation factor a-like effector c (CIDEC)8 and lastly perilipin 1 (PLIN1),9 the latter two involved in lipid droplet formation.10
FPLD type 3 (FPLD3) is a very rare form of lipodystrophy accounting for only approximately 30 patients.5 It results from mutations on the PPARG gene. PPARG is a member of the ligand inducible nuclear transcription factor family expressed in adipose tissue. It is involved in adipogenesis regulation.11 Dominant negative or haploinsufficiency PPARG mutations may inhibit adipocyte differentiation, leading to peripheral subcutaneous fat loss from the arms and legs. Generally, the truncal region, neck and face are spared.6 The age of onset and pattern of progression of fat loss is variable. Similarly to other forms of lipodystrophies, FPLD3 is characterized by a cluster of metabolic disorders, including insulin resistance and hypertriglyceridemia, which subsequently increase the risk of recurrent episodes of acute pancreatitis. Although the exact mechanisms by which PPARG mutations cause insulin resistance remain elusive, alteration in PPARG expression seems to result in adipogenesis deregulation, leading to subcutaneous adipose tissue loss. Thus, the inability to appropriately expand subcutaneous fat mass may cause ectopic triglyceride deposition in liver and muscle, inducing insulin resistance.12,13
We report here a case of a 60-year old Caucasian woman, the child of non-consanguineous, healthy parents. Her neonatal period and psychomotor development were unremarkable. She was referred for endocrinology consultation for dyslipidemia and diabetes with poor glycemic control. She had a past medical history of primary sterility due to bilateral fallopian tube obstruction, diagnosed at the age of 25, and hypertension, since the age of 43. At the age of 45, she presented eruptive xanthoma (Figure 1) and type V dyslipidemia and diabetes were diagnosed. Laboratory work at the first evaluation in our department revealed glycated hemoglobin (HbA1c) of 8.2%, high total plasmatic cholesterol levels (623.9 mg/dL), low plasmatic high-density lipoprotein (HDL) cholesterol levels (9.1mg/dL) and hypertriglyceridemia (triglycerides of 6092 mg/dL). At the time she was medicated with metformin 1000mg q.d., insulin glargine 50IU at breakfast and 50IU at supper, insulin lispro 20IU at breakfast, 20IU at lunch and 20IU at dinner, adjusted according to capillary blood glucose levels. She was also under rosuvastatin 10mg q.d., acetylsalicylic acid 100mg q.d. and losartan 100mg q.d. Three years after, she was hospitalized for acute pancreatitis.
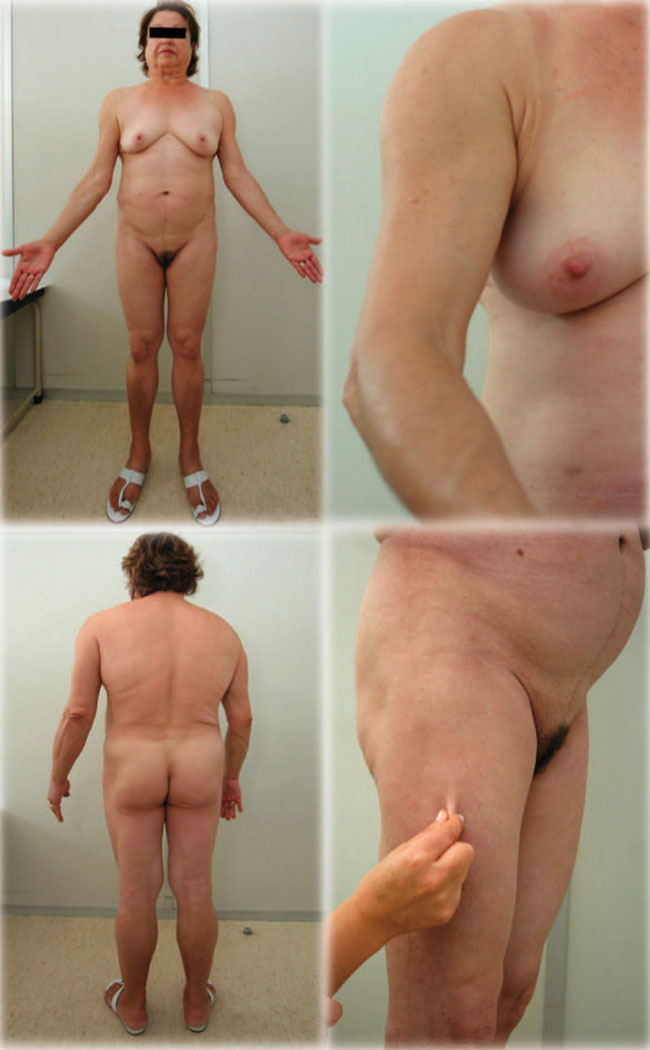
Figure 1. Patients’ physical examination showing moon facies, lipoatrophy of upper and lower limbs and gluteal region and abdominal proeminence.
A clinical interview revealed that the patient had had progressive upper and lower limbs and gluteal region fat loss, accompanied by abdominal fat hypertrophy since the age of 30. She had no family history of lipodystrophy. Physical examination demonstrated fat redistribution, namely moon facies, extremities fat tissue atrophy and abdominal fat prominence (Figure 2). No buffalo hump, hirsutism, acne or achantosis nigricans were found. She was overweight (BMI: 27kg/m2). The genitalia were normal and no apparent hypertrophy was present. Abdominal ultrasound revealed severe hepatic steatosis. She has no target organ damage for diabetes. The last analyses showed a HbA1c of 7.2%, 99 mg/dL of total cholesterol, 28 mg/dL of HDL cholesterol, 398 mg/dL of triglycerides, normal apolipoprotein A1 (109mg/dL) and decreased apolipoprotein B levels [48 mg/dL (normal range 53-138)], under restricted diet, insulin glargine (50IU at breakfast and 50IU at supper) and insulin lispro (insulin lispro 20IU at breakfast, 20IU at lunch, 20IU at snack and 20IU at dinner, adjusted according to capillary blood glucose levels), rosuvastatin 20 mg qd and fenofibrate 267mg qd. To characterize fat mass pattern, a dual-energy x-ray absorptiometry (DEXA) was performed, which confirmed the previously described abnormal adipose tissue distribution, characterized by increased fat tissue of the abdominal region and decreased fat mass in the lower and upper limbs. Fat mass ratio (FMR), determined by the ratio between the percent of the trunk fat mass and the percent of the lower-limb fat mass,14 was 1.44 (24.4/16.9).

Figure 2. Eruptive xanthoma in posterior leg -rash pink papules (firm pinhead-sized bumps) with a creamy center.
Given the suspicion of a partial familial lipodystrophy, a LMNA gene analysis was carried out, which ruled out a possible mutation in that gene, thereby excluding diagnosis of familial partial lipodystophy of Dunningan. Subsequently, a genetic study was conducted to investigate a possible PPARG mutation, leading to identification of a mutation on exon 4 of the PPARG gene (p.Gly161Val; c.482G>T), a variant that has not been previously described, but very likely to be disease-causing, as predicted by two different bioinformatics tools. The identification of this new mutation enabled the diagnosis of familial partial lipodistrophy type 3. (The laboratory PPARG gene analysis was performed at the Novogenia GmbH laboratory.)
The patient’s overall clinic and metabolic phenotype revealed a FPLD form. As functional signs in FPLD are extremely rare, the suspicion of a lipodystophy syndrome requires careful examination and investigation. Therefore, the patient’s interview must assess age of onset of fat mass loss, adipose tissue distribution, clinical features and known metabolic impairments, as well as potential family history of lipodystrophy. Physical examination requires anthropometry evaluation and almost always reveals a body mass index inferior to 25 Kg/m2.15 Photos can also be important to evaluate the progression of lipodystrophy. Although not strictly necessary, DEXA, computed tomography and magnetic resonance imaging can accurately characterize fat mass pattern, particularly in atypical phenotypes.
Dunnigan-type familial partial lipodystrophy is the most frequent form of genetic partial lipodystrophy.16 However, sequencing of the LMNA gene ruled out this type in our patient. Subsequently, a PPARG genetic study enabled identification of a new mutation on c.482G>T (p.Gly161Val), yielding the diagnosis of FPLD3. PPARG is a known key mediator of adipogenesis and fat tissue remodeling, thus regulating insulin sensitivity.17 The deletion of adipose tissue-specific PPARG in mice leads to congenital and progressive lipodystrophy, resulting in free-fatty-acid accumulation in non-adipose tissues, such as skeletal muscle, liver and pancreas.18 Consequently, lipotoxicity induces an insulin resistance state.19 Gradual fat loss, insulin resistance and hypertriglyceridemia are, therefore, common features of this syndrome.20-23
Regarding therapeutic approaches for FPLD3, management of associated metabolic disorders includes personal lifestyle, diet and physical activity measures, along with glycemic control as well as dyslipidemia and hypertension therapy. Thiazolidinediones appear to be a promising therapeutic option as they improve insulin resistance and stimulate differentiation and growth of adipocytes. Unfortunately, they exacerbate fat accumulation in non-affected regions, which limits their administration.24 Metreleptin, a recombinant analogue of human leptin, is a valid option and has been recently approved for the associated metabolic complications of lipodystrophies, including insulin resistance, hyperglycemia and dyslipidemia.25 Concerning cosmetic implications of fat loss, as it is not possible to reverse adipose tissue atrophy, plastic surgery is the only option to improve cosmetic disfigurement.26
The case report aims to highlight the importance of clinical suspicion and early diagnosis of lipodystrophies, as they are closely associated with metabolic complications. Correct identification, prompt and adequate intervention and close long-term follow-up may delay the onset of metabolic disorders and associated cardiovascular disease.
DISCLOSURE
There is no conflict of interest that would undermine the impartiality of this scientific work. There is no potential conflict of interest concerning any author of the manuscript.
REFERENCES
1. Garg A, Misra A, 2004 Lipodystrophies: rare disorders causing metabolic syndrome. Endocrinol Metab Clin North Am 33: 305-331.
2. Garg A, Agarwal AK, 2009 Lipodystrophies: Disorders of adipose tissue biology. Biochim Biophys Acta 1791: 507-513.
3. Haque W, Shimomura I, Matsuzawa Y, Garg A, 2002 Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab 87: 2395-2398.
4. Peters JM, Barnes R, Bennett L, Gitomer WM, Bowcock AM, Garg A, 1998 Localization of the gene for familial partial lipodystrophy (Dunnigan variety) to chromosome 1q21–22. Nat Genet 18: 292-295.
5. Garg A, 2011 Lipodystrophies: Genetic and Acquired Body Fat Disorders. J Clin Endocrinol Metab 96: 3313-3325.
6. Agarwal A, Garg A, 2002 A Novel Heterozygous Mutation in Peroxisome Proliferator-Activated Receptor-γ Gene in a Patient with Familial Partial Lipodystrophy. J Clin Endocrinol Metab 87: 1408-1411.
7. George S, Rochford JJ, Wolfrum C, et al, 2004 A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 304: 1325-1328.
8. Rubio-Cabezas O, Puri V, Murano I, et al, 2009 Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med 1: 280-287.
9. Gandotra S, Le Dour C, Bottomley W, et al, 2011 Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med 364: 740 -748.
10. Olofsson SO, Boström P, Andersson L, et al, 2008 Triglyceride containing lipid droplets and lipid droplet-associated proteins. Curr Opin Lipidol 19: 441-447.
11. Jeninga E, Kalkhoven E, 2010 Central players in inherited lipodystrophies. Trends Endocrinol Metab 21: 581-588.
12. Garg A, 2004 Acquired and inherited lipodistrophies. N Engl J Med 350: 1220-1234.
13. Tan G, Savage D, Fielding B, et al, 2008 Fatty acid metabolism in patients with ppargamma mutations. J Clin Endocrinol Metab 93: 4462-4470.
14. Freitas P, Santos AC, Carvalho D, et al, 2010 Fat mass ratio: an objective tool to define lipodystrophy in HIV-infected patients under antiretroviral therapy. J Clin Densitom 13: 197-203.
15. Vantyghem MC, Balavoine AS, Douillard C, et al, 2012 How to diagnose a lipodystrophy syndrome. Ann Endocrinol (Paris) 73: 170-189.
16. Fardet L, Vigouroux C, Capeau J, 2013 Syndromes lipodystrophiques. Rev Med Interne 34: 614-622.
17. Semple R, Chatterjee V, Rahilly S, 2006 PPAR-gamma and human metabolic disease. J Clin Invest 116: 581-589.
18. He W, Barak Y, Hevener A, et al, 2003 Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A 100: 15712-15717.
19. Medina-Gomes G, Gray S, Vidal-Puig A, 2007 Adipogenesis and lipotoxicity: role of peroxisome proliferator- activated receptor g (PPARg) and PPARgcoactivator-1 (PGC1). Public Health Nutrition 10: 1132-1137.
20. Hegele R, Joy T, Al-Attar D, Rutt B, 2007 Lipodystrophies: windows on adipose biology and metabolism. J Lipid Res 48: 1433-1444.
21. Visser M, Kropman E, Kranendonk, et al, 2011 Characterization of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPAR-gamma mutation (Y151C). Diabetologia 54: 1639-1644.
22. Semple R, Savage D, Cochran E, Gorden P, O’Rahilly S, 2011 Genetic syndromes of severe insulin resistance. Endocr Rev 32: 498-514.
23. Monajemi H, Zhang L, Li G, et al, 2007 Familial Partial lipodistrophy phenotype resulting from a single-base mutation in deoxyribonucleic acid-binding domain of peroxisome proliferator-activated receptor-gamma. J Clin Endocrinol Metab 92: 1606-1612.
24. Walker UA, Kirschfink M, Peter HH, 2003 Improvement of acquired partial lipodystrophy with rosiglitazone despite ongoing complement activation. Rheumatology 42: 393-394.
25. Chong AY, Lupsa BC, Cochran EK, Gorden P, 2010 Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 53: 27-35.
26. Fiorenza C, Chou S, Mantzoros C, 2011 Lipodystrophy: pathophysiology and advances in treatment. Nat Rev Endocrinol 7: 137-150.
Address for correspondence:
Eva Lau, Department of Endocrinology, Diabetes and Metabolism, Faculty of Medicine University of Porto, Centro Hospitalar São João, Porto, Portugal E-mail: evalau.med@gmail.com
Received 23-09-2014, Accepted 27-01-2015