Division of Clinical and Molecular Endocrinology, 1st Department of Internal Medicine, AHEPA University Hospital, Aristotle University of Thessaloniki, Greece
Sclerosteosis and Van Buchem disease are two rare bone sclerosing disorders characterized by increased bone mineral density, tall stature and entrapment of cranial nerves due to overgrowth of a highly dense bone. Recent advances in human genetics have revealed the genetic background of these disorders by cloning the SOST gene, which is localized on chromosome region 17q12-q21 and codes for sclerostin. Sclerostin is a protein produced almost exclusively from osteocytes inhibiting bone formation by both osteoblasts and osteocytes. At the molecular level, sclerostin inhibits the Wnt signaling pathway, which plays a critical role in osteoblast development and function. Induced sclerostin deficiency in mice reproduces the bone sclerosing human diseases, while sclerostin excess leads to bone loss and reduced bone strength. The extracellular nature of sclerostin has rendered it a promising target for the development of novel anti-osteoporotic treatment. Otherwise healthy carriers of the SOST mutation present with increased bone mass and low levels of sclerostin in serum in contrast to patients with sclerosteosis, who exhibit undetectable levels, thus pointing to the possibility of titration of sclerostin levels in the circulation. Based on these unique characteristics, human anti-sclerostin antibodies have been developed and tested in ovariectomized rats and monkeys, demonstrating very promising results in bone formation. Clinical phase II and III trials are currently underway thereby translating human genetics to drug development.
Antibodies, Blosozumab, BPS804, Romosozumab, Sclerostin
INTRODUCTION
Bone formation is an essential process of the development of the human body, starting during fetal development and continuing throughout childhood and adolescence as the skeleton grows. This lifelong process is characterized by continuing cycles of bone remodeling, consisting of bone resorption and bone formation, and it is essential for skeletal morphogenesis during growth (modeling) and the repair of micro-damages in adult life (remodeling).1
Tight coordination of osteoblast, osteocyte and osteoclast function is very important for bone homeostasis.2 Abnormalities in bone remodeling due to impaired signaling or malfunction of these cells result in skeletal disorders.
Sclerosteosis and Van Buchem disease are two rare skeletal disorders characterized by generalized bone overgrowth. Recent advances in molecular genetics have revealed that both of them are caused by functional mutations in the SOST gene, which is localized in the 17q12-21 chromosomal region and codes for sclerostin, a glycoprotein that is solely expressed by osteocytes.3-6 Sclerostin acts as a negative regulator of bone formation, through inhibition of the Wnt signaling pathway, which is of critical importance for the development and function of osteoblasts.7 Undetectable or very low levels of sclerostin in these patients result in excessive bone growth and increased bone strength8 (Figure 1).
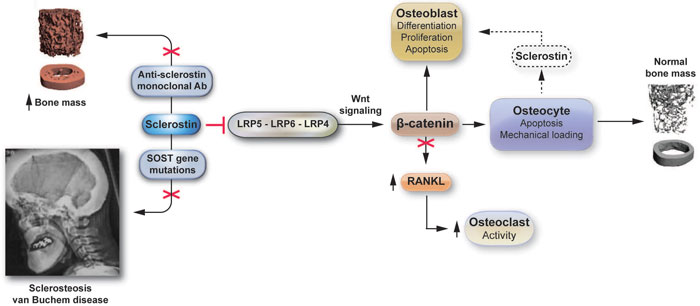
Figure 1. Sclerostin: From genes to antibodies. After its secretion from osteocytes, sclerostin binds to the LRP5/6/4 proteins in the cellular membrane in an autocrine and/or paracrine manner, inhibiting activation of Wnt signaling which regulates differentiation, proliferation and apoptosis of osteoblasts and osteocytes. Sequestration of beta-catenin also leads to activation of osteoclastogenesis. Inactivation of sclerostin either by mutations of the SOST gene or by anti-sclerostin antibodies leads to increased bone formation.
Since the first description of Van Buchem disease and sclerosteosis, several in vitro and in vivo studies have pointed to the critical role of sclerostin in bone remodeling and have paved the way to the development of novel molecular targets for treatment of osteoporosis and other metabolic bone diseases characterized by low bone mass.
SCLEROSING DISORDERS CAUSED BY DYSFUNCTION OF THE SOST GENE
Sclerosteosis
Sclerosteosis is a rare bone dysplasia first described in 1958 and it is most common among Afrikaners, mainly white, of Dutch origin in South Africa, although other cases have also been reported worldwide.9 It is caused by 6 different types of loss-of-function mutations of the SOST gene, resulting in reduced production of sclerostin, and is inherited in an autosomal-recessive mode.6 All patients are homozygous for these mutations and the complete absence of sclerostin results in overgrowth of skull bones, mandible, ribs, clavicles, long bones and pelvis. Patients are usually tall and have a characteristic face because of the facial deformities of bossing of the forehead and enlargement of the mandible.10 In addition, due to narrowing of the foramina of the cranial nerves and the consequent entrapment of nerves, nerve deficits are commonly encountered. Unilateral or bilateral facial nerve paralysis is the most common clinical finding and varies from moderate to severe. Hearing loss and visual impairment have also been reported.11,12 Other malformations include syndactyly, radial deviation of the terminal phalanges and nail dysplasias.13 Moreover, due to cranio-tubular hyperostosis, patients often present with increased intracranial pressure, which is severe and persistent. This results in episodic early morning headaches that are accompanied by nausea and dizziness with the majority of patients requiring surgical intervention during childhood and adolescence.14 In most cases the symptoms appear during early childhood and adolescence and progress until the third decade. After the 25th year of age symptoms usually stabilize with no new clinical findings or progression.8
Interestingly, heterozygote carriers of the SOST mutation have detectable serum sclerostin levels and are symptom-free. However, their bone mass is high normal or high compared to healthy same-aged peers, although lower than in homozygous patients, and they are less prone to fractures. This observation led to the initial hypothesis that there is a gene-dose effect on circulating sclerostin, which results in different clinical phenotypes.8,15
Van Buchem disease
This is a skeletal disorder initially described in 1955, which appears more frequently in persons of Dutch ancestry.16-18 It is caused by a 52-kb deletion downstream of the SOST gene, which results in lack of a SOST-specific regulatory element and, like sclerosteosis, is an autosomal recessive disease.5,19-21 All patients are homozygous for this deletion and have reduced levels of sclerostin, in contrast to sclerosteosis patients who have undetectable levels of sclerostin, and this difference is probably responsible for the milder phenotype of the Van Buchem disease.22 It is characterized by increased bone mass with enlargement of the mandible and macrocephaly but with normal stature. Patients almost never experience fractures, even when involved in major accidents such as falls from heights or car accidents.22 As with sclerosteosis, patients present with complications such as facial palsy, hearing impairment, visual problems, smell deficiency and neuralgic pain due to narrowing of nerve canals and encroachment of nerves on the cranial foramina.22 Similarly, clinical features and complications do not progress after adulthood, but they are less severe because of slower progression of bone overgrowth after the first years of life in comparison to sclerosteosis.3,22 In addition, although not as often as in sclerosteosis, patients have increased intracranial pressure but in most cases they do not need craniotomy. Unlike sclerosteosis, however, Van Buchem patients do not present with syndactyly or nail dysplasias (Table 1).13,22 This is probably explained by the fact that the missing regulatory element of the SOST gene does not control transcription of the SOST gene during the embryonic life.22,23 Heterozygotes are symptom-free and sclerostin levels are lower compared to healthy individuals but are increased compared to homozygous sclerosteotic patients. In addition, the bone mineral density of Van Buchem carriers—some of whom experience fractures—also varies from low to very high, in contrast to sclerosteosis carriers, all of whom present with high normal values.22,24
SCLEROSTIN
Sclerostin is a glycoprotein that belongs to the DAN family of bone morphogenetic protein (BMP) antagonists25 and contains a C-terminal cysteine knot-like domain.25,26 Sclerostin is expressed mainly in osteocytes, and more specifically in mature osteocytes, which are located deeper in the bone, while it is not present in osteocytes near the bone surface.4,26 Due to its similarity to the DAN family of proteins, sclerostin was initially considered to be a BMP antagonist.4 Later studies, however, demonstrated that sclerostin’s mechanism of action is different from that of the classical BMP antagonists and is mediated through inhibition of Wnt signaling activity.4,26
The Wnt signaling pathway is an evolutionary, highly conserved, intracellular signal transduction pathway that participates in various cellular functions in different tissues of the body in a paracrine and/or autocrine manner.26 The signal is transducted through binding of Wnt proteins in a trans-membrane receptor complex that consists of a 7 membrane spanning receptor of the Frizzled family of G-proteins coupled receptors (GPCR) and a single trans-membrane co-receptor that belongs to the low-density lipoprotein family of receptors (LRP5/6/4). Mutations in LRP genes result in a high bone mass phenotype.27
In the absence of Wnt proteins, cytoplasmic beta-catenin is phosphorylated by an intracellular protein-complex, which includes the proteins Disheveled and Axin, and the kinases glycogen synthase kinase 3-beta (GSK3β) and casein kinase (CK). Upon phosphorylation, beta-catenin is degraded by the intracellular proteasome-ubiquitin proteolytic system.26,28 Binding of Wnt proteins to Frizzled receptor triggers the phosphorylation and activation of Disheveled, which in turn causes recruitment of Axin in the membrane where it binds with the intracellular part of the LRP5 receptor and represses GSK-3β activity. This sequence of events allows beta-catenin to escape phosphorylation and translocate into the nucleus, where it binds with TCF/LEF transcription factors and activates transcription of target genes.
Sclerostin is one of the extracellular antagonists of the Wnt signaling pathway, such as Dickkopf, Wnt inhibitory factor (WIF) and secreted Frizzled related proteins (sFRPs). Through an autoregulating mechanism, the transcription of the SOST gene itself is regulated by the canonical Wnt-signaling pathway.26,28
Circulating sclerostin binds to the LRP5/6/4 proteins preventing the intracellular part of LRP5 from restraining Axin and free beta-catenin from the intracellular protein complex, this leading to its phosphorylation and degradation and consequently inhibition of Wnt signaling.
Over the past few years, the Wnt/beta-catenin-signaling pathway has been shown to be an important regulator of bone mass accrual and maintenance by increasing osteoblastogenesis and bone formation. Inactivating mutations in the LRP5 gene result in the osteoporosis pseudoglioma syndrome,29 and gain-of-function mutations give rise to a high bone mass phenotype in humans.30-32 Inactivating mutations in the LRP4 gene result in the Cenani-Lenz syndrome, an autosomal recessive disorder affecting distal limp development.33 The recapitulation of these phenotypes in mice models opened the way to understanding the crucial role of this pathway in bone metabolism.34
REGULATION OF SCLEROSTIN EXPRESSION
Expression of sclerostin by osteocytes is regulated by mechanical forces and hormones that are known to affect bone metabolism such as parathyroid hormone, calcitonin and glucocorticoids.35 Studies in vitro and in animal models have shown that parathyroid hormone inhibits the expression of the SOST gene by osteocytes.36 In line with the in vivo studies, patients with primary hyperparathyroidism due to chronic elevation of PTH have significantly lower serum sclerostin levels compared with patients who have undergone parathyroidectomy and have normal PTH concentrations, thus confirming the down-regulation of the SOST gene by PTH in humans.37 Calcitonin, on the other hand, which inhibits osteoclast resorption, up-regulates sclerostin expression by osteocytes, while it decreases other osteocyte products such as MEPE and DMP.35,38,39
Mechanical stimulation of the skeleton either through exercise or experimental loading induces bone formation, while immobilization increases the number of sclerostin positive osteocytes.40,41 Sclerostin levels are also affected by sex steroids. Estrogen replacement treatment of postmenopausal women inhibits increases of sclerostin levels that normally follow sex steroid deficiency.42 In elderly men, physiologic estrogen prevented the rise of sclerostin levels. On the other hand, testosterone replacement following the GnRH agonist leuprolide acetate suppression of endogenous estrogen and testosterone production did not appear to affect sclerostin levels.42
Glucocorticoids have been shown to increase the expression of sclerostin in vivo and in vitro.43-45 Data in humans are scarce and there is considerable discrepancy between results, probably due to different doses and duration of treatments as well as differences in the characteristics of patients treated with glucocorticoids. In patients with endogenous hypercortisolism, circulating sclerostin levels were reported to be decreased and to increase during biochemical remission of the disease.46 In another study, however, a progressive decrease in Dickkopf-related protein 1 (Dkk-1) serum levels and an increase in circulating sclerostin levels after long-term follow-up have recently been reported in patients treated with high doses of exogenously administered glucocorticoids.47
Apart from circulating hormones, sclerostin expression is also influenced by cytokines acting in the bone microenviroment. Paracrine- and autocrine-acting factors such as prostaglandin E2, oncostatin M, cardiotrophin-1 and osteoblastic transcription factors, including osterix, zinc finger protein 467 and leukemia inhibitory factor, also regulate osteocyte sclerostin expression in a cellular-context dependent manner.48-53
Finally, different stages in osteoblast differentiation can cause changes in sclerostin expression in response to inhibitors of ephrin signaling or to inflammatory mediators such as tumor necrosis factor and tumor necrosis factor-like weak inducer of apoptosis (TWEAK).54,55
SERUM SCLEROSTIN LEVELS
Although serum sclerostin levels have been found to be highly associated with bone marrow levels,56 inconsistent results regarding changes of sclerostin levels in different conditions are frequently encountered. For instance, in some studies,57,58 but not in others,59,60 serum sclerostin levels have been positively associated with increased fracture risk. Regarding bone mass, however, and in contrast to what would normally be expected given that sclerostin is a potent inhibitor of the Wnt signaling pathway, serum sclerostin levels have been found to be positively associated with bone mineral mass. The reason for these findings is not yet clear and probably changes in other parameters of bone metabolism such as bone quality, and bone microarchitecture or other yet unrecognized mechanisms, may contribute. Sclerostin has also been measured in a variety of other clinical conditions including ankylosing spondylitis, chronic kidney disease, diabetes, fractures, multiple myeloma and spinal cord injury.61
Despite increasing knowledge in the field of sclerostin biology, certain issues regarding biological variability and the validity of immunoassay measurement in the serum remain unresolved. Gender, age and seasonal or circadian variability62 are known to affect serum levels of sclerostin. In addition, mechanisms of sclerostin clearance from the circulation either from the liver or kidneys and the potential influence of organ dysfunction in serum measurements as well as the impact of pharmacological intervention63 should also be taken into consideration when interpreting sclerostin levels in clinical practice.
Currently there are three commercial sclerostin assays available, while several studies have reported results from non-commercially available56,57 or laboratory-generated ‘in-house’64 assays. In two of the commercially available assays (Biomedica and TECO assays), a monoclonal anti-sclerostin secondary antibody is used, while in the third assay (MSD assay), which seems to be the only one that detects the intact sclerostin molecule, a polyclonal secondary antibody is in use. Differences between the assays have been reported and probably reflect differences in the epitopes recognized or cross-reactivity with the related sclerostin domain-containing protein 1 (SOST-DC1).65
Taking into consideration the data reported so far, measurements of serum sclerostin levels appear useful for understanding the mechanisms by which osteocytes respond to hormonal, physical and pharmacological stimuli,61 but there are as yet numerous issues to be resolved in order to incorporate these measurements into clinical practice.
Α NOVEL TARGET FOR DRUG DEVELOPMENT IN THE TREATMENT OF OSTEOPOROSIS
Its extracellular nature and its almost exclusive production from osteocytes have rendered sclerostin a promising target for the treatment of osteoporosis. In vivo models of SOST knockout mice and preclinical studies in rodents and primates have confirmed sclerostin’s bone-forming action. Moreover, an interesting observation in humans —the increased bone mass in the absence of symptomatology in heterozygous carriers of SOST inactive mutations8,15— has led to the hypothesis that pharmacological inhibition of sclerostin would not be expected to cause adverse bone-overgrowth effects. Thus a new path for development and use of sclerostin antibodies in humans was opened.
IN VIVO STUDIES
In an in vivo model of sclerostin deficiency, adult SOST knockout mice (SOST-/-) were phenotypically similar to wild type (WT) littermates with normal skeletal appearance,66 in contrast to patients with sclerosteosis who usually present with facial distortions and syndactyly.3,9,13 The only important difference between the two groups of mice was the significantly higher bone mass found in SOST-/- mice. By means of DXA analysis, SOST -/- mice demonstrated significantly higher BMD values (>50%) on both the lumbar vertebrae and the whole leg, compared to wild type and heterozygous for SOST targeted allele mice, whereas there was no significant difference in BMD values between WT and heterozygous mice. Femoral μCT analysis showed significant increases in bone volume of both trabecular and cortical compartments and this was confirmed in histomorphometric analysis where increased osteoblast but not osteoclast surface was apparent in trabecular, endocortical and periosteal surfaces with normal lamellar bone structure in all areas.66 Biologically consistent with the results from the knockout mice were the observations from sclerostin-transgenic mice that overexpressed human SOST gene. As a mirror image of SOST-knockout, these mice exhibited reduced bone mass, disorganized bone microarchitecture, impaired lamellar bone formation and chondrodysplasia.67
PRECLINICAL STUDIES WITH SCLEROSTIN ANTIBODIES
Further investigation into the sclerostin effect on animal bone phenotype was conducted assessing the effectiveness of pharmacological inhibition of sclerostin.
Administration of sclerostin neutralizing monoclonal antibodies was tested in aged ovariectomized rats68 and gonad-intact female cynomolgus monkeys,69 displaying clear anabolic effects with significant increases in bone formation on trabecular, periosteal, endocortical and intracortical surfaces.
In both studies, antibody-mediated sclerostin inhibition resulted in a rapid increase in bone formation that returned to baseline as the antibody was cleared from the circulation. Interestingly, this effect was completely reproducible in sequential administrations, suggesting that similar rapid modulation of bone formation could be expected with pharmacologic regulation of sclerostin activity in humans.
CLINICAL STUDIES WITH SCLEROSTIN ANTIBODIES
In clinical studies, two structurally diverse humanized sclerostin antibodies, romosozumab (AMG 785, CDP-7851; co-developed by Amgen, Thousand Oaks, CA, USA, and UCB, Belgium) and blosozumab (Eli Lilly and Company, Indianapolis, IN, USA) have shown favorable effects in postmenopausal women with osteoporosis, confirming data from preclinical models (Table 2). Τhe first humanized sclerostin monoclonal antibody (romosozumab) was tested in phase 1 and 2 clinical trials demonstrating a well tolerated safety profile and significant gains in bone mass.70,71
In the phase 1 study, romosozumab administered either intravenously (IV) or subcutaneously (SC) in healthy men and postmenopausal women with low bone mass was well tolerated at all doses, exhibiting only mild adverse events (60% for SC romosozumab vs. 64 % for SC placebo and 25% for IV romosozumab vs. 50% for IV placebo), including injection site reactions, back pain, headache, constipation, dizziness and arthralgia.70There was one case in the romosozumab group (10 mg/kg SC) with severe non-specific hepatitis developed one day after dosing that was, however, completely resolved three weeks later (on the 26th day). Mild decreases in ionized calcium were seen in the romosozumab group and were associated with increases in PTH, but this was a transient effect. In this study, romosozumab increased bone formation and decreased bone resorption, leading to significant gains in BMD and thus supporting further clinical investigation.
In the first phase 2 randomized, placebo-controlled parallel group, multicenter study, administration of romosozumab SC at various doses was tested in terms of efficacy and safety over a 12-month trial in approximately 400 postmenopausal women.71
Participants were aged between 55-85 years with lumbar spine, total hip or femoral neck T-score ≤–2.0 and ≥–3.5 and administration of romosozumab was compared to SC placebo, open label alendronate or open label teriparatide.71
The dose of 210 mg of romosozumab monthly significantly increased bone mass at all skeletal sites (11.3% at the spine, 4.1% at the hip and 3.7% at the femoral neck). Moreover, romosozumab induced greater increases in bone mass not only compared to placebo but also compared to alendronate and teriparatide.
Increase in markers of bone formation was reported from the very first week of treatment with romosozumab, reaching maximum levels after one month and decreasing to baseline or lower between the second and the ninth month. The marker of bone resorption beta-crosslaps (β-CTX) in the serum decreased significantly in the first week and remained reduced during the 12-month trial in the romosozumab group. There were no significant differences in the adverse events between the romosozumab and the placebo group except for reactions related to the injection. In particular, the incidence of severe adverse events in the group receiving the highest dose of romosozumab was 10% compared to 14% seen in the placebo group.
At this year’s ASBMR meeting, the researchers McClung and colleagues presented data from the second year of romosozumab treatment followed by one year of denosumab or placebo. Monthly administration of 210 mg romosozumab continued to significantly increase BMD in the lumbar spine (15.7%) and total hip (6%) through the second year, while bone turnover markers procollagen type 1 N-terminal propeptide (P1NP) and β-CTX remained below baseline levels.
Women who switched to denosumab after two years continued to increase BMD at a rate similar to that observed during the second year of treatment with romosozumab. In women who switched to placebo, however, BMD decreased significantly, reaching pretreatment levels at the end of the third year. These results were comparable to those reported in the DATA-Switch study where denosumab prevented bone loss and further increased BMD in women who were pre-treated with teriparatide for two years (results by Benzamin L and colleagues presented at the same ASBMR meeting in 2014).
Computed tomography scans for L1 vertebrae and high resolution quantitative computed tomography (HR-qCT) scans of T12 vertebral bodies were performed in a subset of women treated with romosozumab in order to evaluate the effect of treatment on cortical and trabecular compartment parameters; these results were also presented by Whitmarsh T et al and Damm T et al at this year’s ASBMR meeting. Twelve months’ treatment with romosozumab significantly improved cortical parameters, such as cortical BMD and cortical bone mineral content (BMC) compared to teriparatide or placebo, while trabecular BMD changes were similar between the teriparatide and the romosozumab group. In addition, using HR qCT, researchers were able to demonstrate that gains in the cortical compartment were attributed to both endocortical and periosteal bone matrix apposition, indicating a favorable effect of romosozumab not only on bone mass but also on bone strength.
The clinical effect of romosozumab to reduce fracture risk is currently being evaluated in an ongoing phase 3 pivotal placebo-controlled fracture study in postmenopausal women with osteoporosis (FRAME study, www.clinicaltrials.gov), which is projected to be completed in December 2016.
Safety and tolerability of blosozumab was recently reported in the first phase 1, randomized, placebo-controlled study with escalating doses, single or multiple, for eight weeks.72 There was a change of up to 3.41% (p<0.002) and up to 7.71% (p<0.001) from baseline in lumbar spine BMD at day 85 after single or multiple administrations of blosozumab, respectively, in healthy postmenopausal women aged between 45 and 80 years.
Prior bisphosphonate use did not appear to have an impact on the effects of single doses of blosozumab in terms of changes in bone turnover markers and BMD responses. Blosozumab was generally well tolerated, and the most commonly seen adverse events, regardless of causality, included arthralgias, back pain, fatigue, headache, injection site reactions, nausea and vomiting and respiratory track infections. A dose-related trend towards increases in serum iPTH was associated with a trend towards decrease in urinary and serum calcium. Development of antibodies against blosozumab were detected in 12% of patients after a single dose and in 36% of patients after multiple doses, without, however, evidence of a neutralizing effect or an impact on safety profile.72
Data from a phase 2, randomized, parallel-design, double-blind, placebo-controlled, study that assessed the safety and efficacy of different blosozumab doses given SC in postmenopausal women (n=120) with a mean age 65.8y and mean lumbar spine T-score -2.8, were also recently reported.73 Blosozumab significantly caused dose-related increases in BMD in the lumbar spine, total hip and femoral neck compared to placebo at 52 weeks of treatment. At the highest dose of 270 mg every two weeks, BMD increased 17.7% at the spine and 6.2% at the total hip. Bone formation markers increased rapidly during treatment and decreased towards baseline levels by the end of the study, except for bone-specific alkaline phosphatase that remained higher compared to placebo with the dose of 270 mg every two weeks. The bone resorption marker β-CTX decreased significantly in the first two weeks and remained reduced throughout blosozumab treatment. The researchers demonstrated no significant differences in the adverse events seen between blosozumab treatment and placebo, except for mild injection site reactions that were more frequently encountered with blosozumab (22.6% to 40% vs. 10.3% in the placebo group). These reactions, however, were not associated with the development of anti-drug antibodies. Thirty-five percent of the participants developed anti-drug antibodies but only in one case was a neutralizing effect to blosozumab confirmed.73
Data from a 52-week follow-up of this phase 2 study, where participants did not receive blosozumab, were presented at this year’s ASBMR meeting by Benson C et al. Treatment discontinuation led to decreases in BMD in all blosozumab groups, although for the doses of 270 mg and 180 mg every two weeks, lumbar spine, total hip and femoral neck BMD and lumbar spine and total hip BMD, respectively, remained significantly greater compared to placebo.
Surprisingly, with both antibodies the pattern of anabolic action, as reflected by changes in bone formation markers, was brief and returned to baseline values despite continued administration of the drug, raising questions as to whether this bone anabolic effect will be sustained after prolonged administration (>1 year) or after discontinuation of treatment.74 Even more surprising, however, was the chronic suppression of bone resorption markers. This pattern of bone turnover markers was not seen before with currently available anti-osteoporotic therapies. Potent anti-resorptive agents, such as bisphosphonates, denosumab or cathepsin K inhibitors, suppress both bone formation and bone resorption markers, while teriparatide increases levels of bone-formation markers but, after a delay, increases bone-resorption markers as well. A plausible explanation for the suppressive effect of sclerostin antibodies on bone resorption could be the decrease of RANKL production as well as the increase of osteoprotegerin by the osteocyte.74 Nevertheless, results regarding sustained efficacy and safety from longer administration (>1 year) of sclerostin antibodies are not yet available, thus the optimal duration of treatment which is associated with the highest rate of response is currently unknown.75 Ongoing phase 3 clinical trials with romosozumab will probably yield some answers.
In line with the above, the efficacy and safety of a third IgG2 humanized sclerostin antibody, BPS804 (Novartis, Basel, Switzerland) (Table 2), has been evaluated in a randomized, double-blind, placebo-controlled phase II study [ClinicalTrials.gov identifier: NCT01406548]. The study was completed in October 2013 but the results were not yet announced at the time of writing. The primary efficacy endpoint is change in lumbar spine BMD at month 9 compared with baseline. Subjects for this study are postmenopausal women aged 45-85 years with a baseline lumbar spine T score from –2 to –3.5.
Safety and tolerability of BPS804 is also being evaluated in adults with hypophosphatasia [ClinicalTrials. gov identifier: NCT01406977] and osteogenesis imperfecta [ClinicalTrials. gov identifier: NCT01417091].
CONCLUDING REMARKS
The sclerostin journey is an exciting one, progressing from human genetics to the development of novel anabolic treatment for osteoporosis. The challenging initial studies on sclerosteosis and Van Buchem disease have stimulated efforts and paved the way for more specific, advanced and targeted research into the underlying genetic deficiency.
Antibody-mediated inactivation of sclerostin appears to be a very promising therapeutic approach demonstrating a favorable safety profile and significant gains in the restoration of bone mass. The restricted expression of the SOST gene in skeletal tissue points to a low risk of extra-skeletal effects in line with the safety data presented so far by the two available sclerostin antibodies.
However, controversy already exists regarding the potential effect of sclerostin on the vasculature,76 since expression of SOST protein has been documented in calcifying vascular tissues in vitro70,71 as well as in clinical studies and Wnt signaling plays a critical role in various aspects of vascular pathophysiology.77
In hemodialysis patients, serum sclerostin levels were found to be inversely associated with mortality,78 raising concerns about the long-term cardiovascular safety of sclerostin inhibition. Further investigation is needed in order to address this critical issue and shed more light on the potential effects of sclerostin on the vasculature.
Despite concerns, however, anti-sclerostin therapy appears to be a potential strategy for the management of osteoporosis and other skeletal disorders and clinical phase II and III studies assessing fracture risk are ongoing, completing the journey from human genetics to drug development (Table 3).
REFERENCES
1. Manolagas SC, Jilka RL, 1995 Bone marrow, cytokines, and bone remodeling. Emerging insights into the pathophysiology of osteoporosis. N Engl J Med 332: 305-311.
2. Manolagas SC, 2000 Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 21: 115-137.
3. Beighton P, Durr L, Hamersma H, 1976 The clinical features of sclerosteosis. A review of the manifestations in twenty-five affected individuals. Ann Intern Med 84: 393-397.
4. van Bezooijen RL, Roelen BA, Visser A, et al, 2004 Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199: 805-814.
5. Balemans W, Ebeling M, Patel N, et al, 2001 Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 10: 537-543.
6. Piters E, Culha C, Moester M, et al, 2010 First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum Mutat 31: E1526-1543.
7. van Bezooijen RL, Svensson JP, Eefting D, et al, 2007 Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res 22: 19-28.
8. van Lierop AH, Hamdy NA, Hamersma H, et al, 2011 Patients with sclerosteosis and disease carriers: human models of the effect of sclerostin on bone turnover. J Bone Miner Res 26: 2804-2811.
9. Beighton P, 1988 Sclerosteosis. J Med Genet 25: 200-203.
10. Hamersma H, Gardner J, Beighton P, 2003 The natural history of sclerosteosis. Clin Genet 63: 192-197.
11. Booth JB, 1982 Medical management of sensorineural hearing loss. Part II: Musculo-skeletal system. J Laryngol Otol 96: 773-795.
12. Tacconi P, Ferrigno P, Cocco L, et al, 1998 Sclerosteosis: report of a case in a black African man. Clin Genet 53: 497-501.
13. Beighton P, Barnard A, Hamersma H, van der Wouden A, 1984 The syndromic status of sclerosteosis and van Buchem disease. Clin Genet 25: 175-181.
14. du Plessis JJ, 1993 Sclerosteosis: neurosurgical experience with 14 cases. J Neurosurg 78: 388-392.
15. Gardner JC, van Bezooijen RL, Mervis B, et al, 2005 Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab 90: 6392-6395.
16. Van Buchem FS, Hadders HN, Ubbens R, 1955 An uncommon familial systemic disease of the skeleton: hyperostosis corticalis generalisata familiaris. Acta radiol 44: 109-120.
17. Van Buchem FS, Hadders HN, Hansen JF, Woldring MG, 1962 Hyperostosis corticalis generalisata. Report of seven cases. Proc K Ned Akad Wet C 65: 205-217.
18. van Buchem FS, 1971 Hyperostosis corticalis generalisata. Eight new cases. Acta Med Scand 189: 257-267.
19. Brunkow ME, Gardner JC, Van Ness J, et al, 2001 Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet 68: 577-589.
20. Staehling-Hampton K, Proll S, Paeper BW, et al, 2002 A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 110: 144-152.
21. Balemans W, Patel N, Ebeling M, et al, 2002 Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 39: 91-97.
22. van Lierop AH, Hamdy NA, van Egmond ME, Bakker E, Dikkers FG, Papapoulos SE, 2013 Van Buchem disease: clinical, biochemical, and densitometric features of patients and disease carriers. J Bone Miner Res 28: 848-854.
23. Loots GG, Kneissel M, Keller H, et al, 2005 Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res 15: 928-935.
24. Vanhoenacker FM, Balemans W, Tan GJ, et al, 2003 Van Buchem disease: lifetime evolution of radioclinical features. Skeletal Radiol 32: 708-718.
25. Lewiecki EM, 2011 Sclerostin: a novel target for intervention in the treatment of osteoporosis. Discov Med 12: 263-273.
26. Yavropoulou MP, Yovos JG, 2007 The role of the Wnt signaling pathway in osteoblast commitment and differentiation. Hormones (Athens) 6: 279-294.
27. Johnson ML, 2004 The high bone mass family--the role of WntLrp5 signaling in the regulation of bone mass. J Musculoskelet Neuronal Interact 4: 135-138.
28. Ott SM, 2005 Sclerostin and Wnt signaling--the pathway to bone strength. J Clin Endocrinol Metab 90: 6741-6743.
29. Gong Y, Slee RB, Fukai N, et al, 2001 LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107: 513-523.
30. Little RD, Carulli JP, Del Mastro RG, et al, 2002 A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet 70: 11-19.
31. Boyden LM, Mao J, Belsky J, et al, 2002 High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med 346: 1513-1521.
32. Van Wesenbeeck L, Cleiren E, Gram J, et al, 2003 Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet 72: 763-771.
33. Li Y, Pawlik B, Elcioglu N, et al, 2010 LRP4 mutations alter Wntbeta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am J Hum Genet 86: 696-706.
34. Kato M, Patel MS, Levasseur R, et al, 2002 Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol 157: 303-314.
35. Sims NA, Chia LY, 2012 Regulation of sclerostin expression by paracrine and endocrine factors. Clin Rev Bone Miner Metab 10: 98-107.
36. Bellido T, Ali AA, Gubrij I, et al, 2005 Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146: 4577-4583.
37. van Lierop AH, Witteveen JE, Hamdy NA, Papapoulos SE, 2010 Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur J Endocrinol 163: 833-837.
38. Gooi JH, Pompolo S, Karsdal MA, et al, 2010 Calcitonin impairs the anabolic effect of PTH in young rats and stimulates expression of sclerostin by osteocytes. Bone 46: 1486-1497.
39. Robling AG, Niziolek PJ, Baldridge LA, et al, 2008 Mechanical stimulation of bone in vivo reduces osteocyte expression of Sostsclerostin. J Biol Chem 283: 5866-5875.
40. Bonnet N, Ferrari SL, 2010 Exercise and the skeleton: how it works and what it really does. IBMS BoneKEy 7: 235-248.
41. Gaudio A, Pennisi P, Bratengeier C, et al, 2010 Increased sclerostin serum levels associated with bone formation and resorption markers in patients with immobilization-induced bone loss. J Clin Endocrinol Metab 95: 2248-2253.
42. Modder UI, Clowes JA, Hoey K, et al, 2011 Regulation of circulating sclerostin levels by sex steroids in women and in men. J Bone Miner Res 26: 27-34.
43. Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE, 2008 Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum 58: 1674-1686.
44. Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R, 2005 Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun 329: 177-181.
45. Guanabens N, Gifre L, Peris P, 2014 The role of Wnt signaling and sclerostin in the pathogenesis of glucocorticoid-induced osteoporosis. Curr Osteoporos Rep 12: 90-97.
46. van Lierop AH, van der Eerden AW, Hamdy NA, Hermus AR, den Heijer M, Papapoulos SE, 2012 Circulating sclerostin levels are decreased in patients with endogenous hypercortisolism and increase after treatment. J Clin Endocrinol Metab 97: E1953-1957.
47. Gifre L, Ruiz-Gaspa S, Monegal A, et al, 2013 Effect of glucocorticoid treatment on Wnt signalling antagonists (sclerostin and Dkk-1) and their relationship with bone turnover. Bone 57: 272-276.
48. Genetos DC, Yellowley CE, Loots GG, 2011 Prostaglandin E2 signals through PTGER2 to regulate sclerostin expression. PLoS One 6: e17772.
49. Walker EC, McGregor NE, Poulton IJ, et al, 2008 Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res 23: 2025-2032.
50. Walker EC, McGregor NE, Poulton IJ, et al, 2010 Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest 120: 582-592.
51. Yang F, Tang W, So S, de Crombrugghe B, Zhang C, 2010 Sclerostin is a direct target of osteoblast-specific transcription factor osterix. Biochem Biophys Res Commun 400: 684-688.
52. Quach JM, Walker EC, Allan E, et al, 2011 Zinc finger protein 467 is a novel regulator of osteoblast and adipocyte commitment. J Biol Chem 286: 4186-4198.
53. Sims NA, Johnson RW, 2012 Leukemia inhibitory factor: a paracrine mediator of bone metabolism. Growth Factors 30: 76-87.
54. Lorenzo J, 2008 Ephs and ephrins: a new way for bone cells to communicate. J Bone Miner Res 23: 1168-1169.
55. Vincent C, Findlay DM, Welldon KJ, et al, 2009 Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res 24: 1434-1449.
56. Drake MT, Srinivasan B, Modder UI, et al, 2010 Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J Clin Endocrinol Metab 95: 5056-5062.
57. Arasu A, Cawthon PM, Lui LY, et al, 2012 Serum sclerostin and risk of hip fracture in older Caucasian women. J Clin Endocrinol Metab 97: 2027-2032.
58. Ardawi MS, Rouzi AA, Al-Sibiani SA, Al-Senani NS, Qari MH, Mousa SA, 2012 High serum sclerostin predicts the occurrence of osteoporotic fractures in postmenopausal women: the Center of Excellence for Osteoporosis Research Study. J Bone Miner Res 27: 2592-2602.
59. Garnero P, Sornay-Rendu E, Munoz F, Borel O, Chapurlat RD, 2013 Association of serum sclerostin with bone mineral density, bone turnover, steroid and parathyroid hormones, and fracture risk in postmenopausal women: the OFELY study. Osteoporos Int 24: 489-494.
60. Szulc P, Bertholon C, Borel O, Marchand F, Chapurlat R, 2013 Lower fracture risk in older men with higher sclerostin concentration: a prospective analysis from the MINOS study. J Bone Miner Res 28: 855-864.
61. Clarke BL, Drake MT, 2013 Clinical utility of serum sclerostin measurements. Bonekey Rep 2: 361.
62. Dawson-Hughes B, Harris SS, Ceglia L, Palermo NJ, 2014 Serum sclerostin levels vary with season. J Clin Endocrinol Metab 99: E149-152.
63. Gatti D, Viapiana O, Adami S, Idolazzi L, Fracassi E, Rossini M, 2012 Bisphosphonate treatment of postmenopausal osteoporosis is associated with a dose dependent increase in serum sclerostin. Bone 50: 739-742.
64. Appel H, Ruiz-Heiland G, Listing J, et al, 2009 Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheum 60: 3257-3262.
65. He JW, Yue H, Hu WW, Hu YQ, Zhang ZL, 2011 Contribution of the sclerostin domain-containing protein 1 (SOSTDC1) gene to normal variation of peak bone mineral density in Chinese women and men. J Bone Miner Metab 29: 571-581.
66. Li X, Ominsky MS, Niu QT, et al, 2008 Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. Journal of Bone and Mineral Research 23: 860-869.
67. Winkler DG, Sutherland MK, Geoghegan JC, et al, 2003 Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22: 6267-6276.
68. Li X, Ominsky MS, Warmington KS, et al, 2009 Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24: 578-588.
69. Ominsky MS, Vlasseros F, Jolette J, et al, 2010 Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res 25: 948-959.
70. Padhi D, Allison M, Kivitz AJ, et al, 2013 Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: A randomized, double-blind, placebo-controlled study. J Clin Pharmacol: doi: 10.1002jcph.239.
71. McClung MR, Grauer A, Boonen S, et al, 2014 Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370: 412-420.
72. McColm J, Hu L, Womack T, Tang CC, Chiang AY, 2014 Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J Bone Miner Res 29: 935-943.
73. Recker R, Benson C, Matsumoto T, et al, 2014 A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res: doi: 10.1002jbmr.2351.
74. Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ, 2011 Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One 6: e25900.
75. Becker CB, 2014 Sclerostin inhibition for osteoporosis--a new approach. N Engl J Med 370: 476-477.
76. Evenepoel P, D’Haese P, Brandenburg V, 2014 Romosozumab in postmenopausal women with osteopenia. N Engl J Med 370: 1664.
77. Bostrom KI, Rajamannan NM, Towler DA, 2011 The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res 109: 564-577.
78. Viaene L, Behets GJ, Claes K, et al, 2013 Sclerostin: another bone-related protein related to all-cause mortality in haemodialysis? Nephrol Dial Transplant 28: 3024-3030.
Address for correspondence:
Maria P. Yavropoulou, MD, MSc, PhD, Division of Clinical and Molecular Endocrinology, AHEPA Univ. Hospital, 1 S. Kyriakidi Str., 54636, Thessaloniki, Greece, Tel.: +302310 993187, Fax: +302310 994608, E-mail: margia@med.auth.gr
Received: 29-02-2014, Accepted: 03-11-2014