Centre for Endocrinology, William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, Charterhouse Square, London, UK
Ghrelin, an orexigenic protein with a unique lipid chain modification, is considered to be an important gut-brain signal for appetite control and energy balance. The ghrelin receptor, growth-hormone secretagogue receptor type 1a, is able to bind acylated ghrelin. The first recognised effect of ghrelin was the induction of growth hormone release from the somatotroph cells of the anterior pituitary. Moreover, by acting on vagal afferents or centrally, ghrelin can activate hypothalamic arcuate neurons that secrete the orexigenic peptides neuropeptide Y and agouti-related peptide, and inhibit the anorexigenic neurons secreting pro-opiomelanocortin and α-melanocyte-stimulating hormone. The orexigenic signalling pathway of ghrelin involves adenosine monophosphate-activated protein kinase. It has been proposed that ghrelin can also increase dopaminergic transmission from the ventral tegmental area to the nucleus accumbens, leading to augmentation of afferent reward signals. Present evidence suggests that ghrelin plays an important role in obesity, eating disorders, and cachexia, as well as in regulating appetite and energy balance in healthy individuals. In pathological states, ghrelin can be lower than normal as is seen in obese individuals, or can be higher than normal as has been reported for Prader-Willi syndrome, anorexia nervosa, bulimia nervosa, and certain types of cachexia. In the future, the application of compounds targeting the ghrelin pathway could involve the use of pharmacotherapies of ghrelin agonists, antagonists or inverse agonists, neutralisation of ghrelin by vaccines and spiegelmers, desacyl ghrelin analogues, as well as inhibitors of the GOAT enzyme which attaches the lipid modification to desacyl ghrelin to synthetise ghrelin.
Anorexia, Ghrelin, GHSR, Obesity, Prader-Willi syndrome
INTRODUCTION
Ghrelin is the only known peripherally produced and centrally active orexigenic hormone that is considered to be an important gut-brain signal for appetite control and energy balance. Its first recognised action was stimulation of growth hormone (GH) release from the pituitary gland but it also stimulates prolactin and ACTH and may inhibit LH/FSH. In addition to its orexigenic and pituitary activity, ghrelin acts on various systems, such as the gastrointestinal, cardiovascular, pulmonary, reproductive, and central nervous systems. The muscle effects of ghrelin involve stimulatory action on the cardiac muscle, vascular smooth muscle, ocular smooth muscle, gastrointestinal smooth muscle, and skeletal muscle and via the GH effect also on bone.1,2 Ghrelin and its receptors are found in the reproductive organs and in the placenta, and during pregnancy and lactation ghrelin regulates maternal energy intake.3 Furthermore, ghrelin could also act as a regulator of the gonadotropic axis.4 It has recently been shown that ghrelin plays a role in the regulation of sodium reabsorption in the cortical collecting ducts of kidneys.5 Ghrelin levels have been found to be altered in various conditions, examples of which are summarised in Table 1. In this paper we focus on the role of ghrelin in obesity, Prader-Willi syndrome, anorexia and other eating disorders, and cachexia, as well as on the implications of ghrelin as a pharmacological target in relation to these disorders.
GHRELIN
Structure, receptor, and signalling pathway
Ghrelin is a 28-amino-acid peptide that is mainly produced by the neuroendocrine cells (named “X/A like” in rat and “P/D1” in humans) in the oxyntic mucosa of the gastric fundus.19 Ghrelin is also produced in areas of the gastrointestinal tract and other peripheral organs but in lesser amounts. The 117-amino-acid preproghrelin protein is encoded by the human ghrelin gene on chromosome 3p25-26. Ghrelin is first cleaved from the preproghrelin polypeptide at the N-terminal by the prohormone convertase 1/3. An octanoic acid then esterifies a hydroxyl group of the third N-terminal amino-acid serine residue of the proghrelin peptide to form acyl ghrelin. Yang et al (2008) demonstrated that the enzyme that attaches octanoate to serine-3 of ghrelin is GOAT (Ghrelin O-Acetyltransferase); this leads to the acylation of ghrelin which is required in order that ghrelin can bind to its receptor.20 However, not all ghrelin is active acylated ghrelin; in fact, only a minority of the circulating ghrelin is acylated. Initially, it was thought that only acyl ghrelin activates the classical growth hormone secretagogue receptor (GHSR) expressing cells, whereas the nonmodified des-n-octanyl form of ghrelin (desacyl ghrelin) does not.21 Meanwhile, although desacyl ghrelin was originally considered an inactive by-product of ghrelin secretion or degradation, recent evidence suggests that desacyl ghrelin acts in peripheral tissues and in the brain to regulate biological actions. A study by Heppner et al (2013) has shown that both acyl ghrelin and desacyl ghrelin significantly increased IP3 formation in HEK-293 cells transfected with human GHSR.22 Intracerebroventricular (icv) infusion of desacyl ghrelin in mice increased fat mass at the highest dose tested, as well as glucose-stimulated insulin secretion. In contrast, icv desacyl ghrelin failed to regulate fat mass and induce hyperinsulinaemia in GHSR deficient (Ghsr-/-) mice. Desacyl ghrelin also failed to regulate these parameters in control mice when it was administered subcutaneously. Based on these findings, the authors suggested that desacyl ghrelin is an agonist of GHSR and regulates body adiposity and peripheral glucose metabolism through a CNS GHSR-dependent mechanism.22
GHSR has two variants: GHSR1a and GHSR1b. GHSR1a is able to bind acylated ghrelin, whereas GHSR1b is physiologically inactive, although one study suggested a dominant-negative, antagonistic role.23 The results of this study showed that when the expression of GHSR1b exceeded that of GHSR1a, there was a decrease in the cell surface expression of GHSR1a with a consequent decrease in constitutive activation of phosphatidylinositol-specific phospholipase C. GHSR1a is a highly conserved G-protein coupled receptor and both GHSR1a and 1b have a wide expression in different tissues, with a role in various systems such as cardiovascular, immune, gastrointestinal, reproductive, and endocrine. GHSR1a is predominantly expressed in the pituitary and at much lower levels in the thyroid gland, pancreas, spleen, myocardium, and adrenal gland.24 Interestingly, the locus of the GHSR gene has been implicated in the determination of height.25
Feedback systems and appetite regulation
The first recognised effect of ghrelin was the induction of GH release from the somatotroph cells of the anterior pituitary. Moreover, by acting centrally or via vagal afferents, ghrelin can activate hypothalamic arcuate neurons that secrete the orexigenic peptides neuropeptide Y (NPY) and agouti-related peptide, and inhibit the anorexigenic neurons secreting α-melanocyte-stimulating hormone (α-MSH). Arcuate neurons project to paraventricular, lateral hypothalamic, and other nuclei, as well as orexin in the lateral hypothalamus, which could contribute to the orexigenic effects of ghrelin.26 Ghrelin is the first known peripheral hormone that can induce orexigenic effects through acting on hypothalamic pathways; moreover, it is active even with peripheral administration, in contrast to most orexigenic peptides which are only active when injected into the brain. Ghrelin is considered to be the peripheral counterpart of insulin and leptin, as it exerts opposite effects compared to these hormones (Figure 1). A study on healthy volunteers demonstrated that ghrelin reduces glucose-stimulated insulin secretion, leading to deterioration of glucose tolerance.28 Moreover, the results of the CODING study demonstrated that a high circulating ghrelin level is associated with lower insulin resistance in the general population.27 In addition, Tong et al showed that physiologic concentrations of exogenously infused ghrelin reduce insulin secretion without affecting insulin sensitivity in healthy humans, indicating that ghrelin antagonism could improve β-cell function.28

Figure 1. Simplified scheme showing that ghrelin stimulates the NPY/AGRP orexigenic pathway and inhibits the POMC-related anorectic pathway, resulting in increased food intake.
Tschop et al demonstrated that peripheral daily administration of ghrelin caused weight gain by reducing fat utilisation in mice and rats, this implying that ghrelin can induce an increase in adipose tissue and body weight.29 Consistent with these results are the findings that ghrelin increases the mRNA expression of fat storage enzymes whilst decreasing the expression of the rate-limiting step in fat oxidation, carnitine palmitoyl transferase-1alpha.30 These results may suggest that ghrelin induces a metabolic switch from the utilisation of fat to carbohydrates, whereas energy expenditure does not appear to be affected.
It has been proposed that ghrelin acts as an initiator signal for food consumption in humans: repeated plasma samples throughout a 24-h period demonstrated a clear preprandial rise and postprandial fall in plasma ghrelin levels, indicating that ghrelin can play a physiological role in meal inititation.31
More recently an important interplay has been demonstrated between glucagon and ghrelin. The mechanisms by which glucagon induces satiety are not completely understood and it has been suggested that glucagon-induced reduction in total ghrelin exerted at the hypothalamo-pituitary level might be responsible for this effect.32
The role of ghrelin in reward-based eating
GHSR1a is expressed in various nodes of the mesolimbic system, which include the ventral tegmental area (VTA) and the nucleus accumbens (NAc). More than 50% of dopaminergic VTA neurons co-express GHSR1a and are also present in GABAergic VTA neuronal subpopulations, which regulate the activity of the dopaminergic neurons.33 It has been proposed that ghrelin can increase the dopaminergic transmission from VTA to the NAc, leading to augmentation of afferent reward signals. A recent study using a rat model demonstrated that VTA injections of ghrelin produced a significant increase in food motivation/reward behaviour, as measured by sucrose-induced progressive ratio operant conditioning, and chow intake. Moreover, pretreatment with either a D1-like or D2 receptor antagonist into the NAc completely blocked the reward effect of ghrelin, leaving chow intake intact.34 These results imply that the VTA to NAc dopaminergic projections, along with D1-like and D2 receptors in the NAc, are essential elements of the ghrelin responsive circuits controlling food reward behaviour.
Homeostatic feeding is thought to be under the control of circulating hormones acting primarily on the hypothalamus. The mechanisms by which ghrelin promotes food intake are not only homeostatic but also include enhancement of the rewarding effects of certain foods so that extra effort is made to obtain the pleasurable food, “hedonic feeding”.35 Hedonic feeding occurs in the absence of nutritional and caloric deficiency and can be described as “non-homeostatic”. Functional magnetic resonance imaging (fMRI) following ghrelin administration demonstrated an increased neural response to food pictures in regions of the brain, including the amygdala, orbitofrontal cortex, anterior insula, and striatum, implicated in encoding the incentive value of food cues.36 The effects of ghrelin on the amygdala and orbitofrontal cortex response were correlated with self-rated hunger ratings.36 The results of this study suggest that metabolic signals such as ghrelin may favour food consumption by enhancing the hedonic and incentive responses to food-related cues.
Several studies have investigated the role of ghrelin in defining food preference. Ghrelin shifts food preference to diets rich in fat29 or sweet food, regardless of whether the food has caloric content. Ghrelin increased the preference for 0.3% saccharin solution in wild type mice, while it did not have an effect in GHSR1 deficient mice, indicating that the ghrelin receptor pathway is necessary to mediate these phenomena.37 In addition to enhancing preference for fatty and sweet foods, ghrelin also mediates more complex reward-based eating behaviours. Some studies have used the conditioned place preference test, which involves comparing the amount of time animals spend in an environment with which they have been conditioned to find a pleasurable diet to the time spent in a distinct environment associated with regular chow or no food. Another method that has been used is operant conditioning, for example operant lever pressing for high-rewarding foods. Studies have demonstrated that by using both the conditioned place preference test and operant conditioning, ghrelin enhanced the rewarding value of a high-fat diet when administered to ad lib-fed mice; by contrast, wild-type mice treated with ghrelin receptor antagonist and ghrelin receptor-null mice both failed to show conditioned place preference test to high-fat diet normally observed under calorie restriction.38,39 Furthermore, use of GHSR1 antagonists reduced operant responding for sucrose solution, as it was found that the intake and self-administration of sucrose in rats was reduced, as was also saccharin intake in mice.40
OBESITY
Various studies have demonstrated that plasma ghrelin levels in obese individuals are low and inversely proportional to body mass index.41 Subjects with insulin resistance and high insulin levels also experience chronically low ghrelin levels. For example, a study analysing the fasting plasma ghrelin concentrations of 1,040 subjects demonstrated that ghrelin concentrations were negatively associated with fasting insulin and insulin resistance.15 Ghrelin concentrations are also different in obese and normal-weight pre-pubertal children: in response to intake of a standardised breakfast, obese children showed an early recovery of postprandial ghrelin levels to baseline levels at 3 hours, while ghrelin levels remained suppressed after 3 hours in normal-weight children.42 In addition to ghrelin levels being low in obese individuals, it has also been suggested that diet-induced obesity causes ghrelin resistance in NPY/AgRP neurons. Diet-induced obesity suppresses the neuroendocrine ghrelin system by decreasing acylated and total plasma ghrelin, decreasing ghrelin and GOAT mRNA in the stomach, decreasing expression of hypothalamic GHSR, and decreasing expression of Npy and Agrp mRNA.43
It has been proposed that genetic polymorphisms of the ghrelin gene may play a role in the relationship of ghrelin and obesity. The Leu72Met change in the preprogherin protein and the Arg51Gln variant at the end of mature ghrelin product (therefore possibly interfering with protein cleavage) has been studied in several cohorts,44-46 but reproducible robust correlations were not found. The GHSR gene has also been studied in obese and diabetic populations47,48 with, however, no consistent results. Interestingly, the GHSR gene has been identified as factor in determining height.25 Rare loss-of-function mutations have been identified in short children. Moreover, Pugliese-Pires et al reported inactivating mutations in the GHSR1 gene in patients with constitutional delay of growth and puberty.49
The role of ghrelin in genetic obesity is an interesting question. Ablation of ghrelin in leptin-deficient ob/ob mice increases insulin secretion and improves hyperglycaemia. Surprisingly however, GHSR ablation worsened the hyperglycaemia, decreased insulin, and impaired glucose tolerance.50 Five human homozygous leptin mutation patients and eight heterozygous carriers were studied for postprandial hormone changes. Plasma ghrelin levels in homozygotes remained remarkably unchanged following food consumption in contrast to a significant decline in heterozygous and normal subjects.51 The effect of MC4R single nucleotide polymorphisms (SNPs) was examined in a large family study; to qualify for the study, families were required to have at least one overweight child between the ages of 4 and 19 years old. The results showed that genetic variation in MC4R plays a functional role in the regulation of physical activity, energy expenditure, and fasting serum ghrelin in Hispanic children; moreover, one of the SNPs (rs34114122) was selected as having likely functional effects on ghrelin, as Bayesian quantitative trait nucleotide analysis strongly supported a functional effect for rs34114122 on fasting serum ghrelin with a strong posterior probability (0.81).52 A link has recently been identified between the obesity-related FTO gene and ghrelin.53 The results demonstrated that the subjects homozygous for the FTO obesity-risk variant had dysregulated circulating levels of acyl ghrelin and attenuated postprandial appetite reduction; moreover, postprandial functional MRI showed that the same subjects had increased neuronal activity in brain regions controlling appetite, reward, and motivation after a test meal. The authors suggested that the FTO genotype can alter the way the brain responds to circulating ghrelin. They moreover proposed a direct link between FTO risk alleles and ghrelin action, as it was found that FTO overexpression in cell-based assays increased ghrelin mRNA expression and total levels of active ghrelin compared with controls, whereas methylation of ghrelin mRNA was reduced. A prospective study used cross-sectional data from 985 elderly patients and demonstrated a positive relationship between the number of FTO C risk alleles (rs17817449) and plasma ghrelin levels; conversely, serum levels of the satiety-enhancing hormone leptin were inversely linked to the number of FTO C risk alleles.54 These associations imply that FTO may facilitate weight gain in humans by shifting the endocrine balance from leptin towards ghrelin.
PRADER-WILLI SYNDROME
The Prader-Willi syndrome (PWS) is the most common cause of syndromic obesity, and results from paternally inherited genes in the q11-13 region of chromosome 15.55 The SNORD116 locus lies in the 15q11-13 region of paternally expressed genes implicated in PWS and it has been proposed that lack of the paternal SNORD116 gene cluster has a determinant role in the pathogenesis of PWS.56 It is characterised by initial poor feeding followed by hyperphagia from the age of 12 months leading to morbid obesity if uncontrolled. Despite their high BMI, several studies found high ghrelin levels in PWS adults and even more in children compared to normal subjects and BMI matched simple obese patients.57-59 These studies might indicate that the hyperphagia observed in PWS could be secondary to elevated ghrelin levels. However, we need to keep in mind that PWS patients are not insulin resistant, rather they have increased insulin sensitivity. A more recent cross-sectional study measuring fasting plasma ghrelin levels in very young children with PWS (n=42) and controls (n=9) aged 7 months to 5 years found no hyperghrelinaemia in young children with PWS, and ghrelin changes were not associated with the timing of the transition to the characteristic hyperphagic phase. The authors suggested that abnormal and/or delayed development or sensitivity of the effector pathways of ghrelin (for example, parasympathetic and central nervous system) may interact with later hyperghrelinaemia to contribute to hyperphagia in PWS.60
In regard to the studies demonstrating elevated ghrelin levels in PWS, several hypotheses have been put forward to account for these observations.61 An increased number of ghrelin-producing cells in the gastric body and fundus of PWS patients could lead to elevated ghrelin levels.62 Imprinting of paternal genes in region q11–13 on chromosome 15 may induce the production of excessive amounts of transcription factors that increase ghrelin expression. There could also be loss of a transcription inhibitory factor that normally suppresses ghrelin expression.63 Reduced visceral adiposity and relative hypoinsulinaemia could partially explain hyperghrelinaemia in PWS.64 Abnormal parasympahetic vagal innervation of the stomach and abnormal sympathetic tone in PWS patients might also explain the high ghrelin levels.65 Moreover, a study has shown that ghrelin dysregulation in PWS occurred very early and preceded the onset of obesity, indicating that in the first years of life, hyperghrelinaemia in PWS may be a response to failure to thrive or food restriction.66 The lack of ghrelin suppression after meals in PWS adults59 but not in PWS children67 may imply that the progressive deterioration of PWS could result in a multistep developmentally driven process in ghrelin dysregulation. Also, in adults with PWS, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-1 levels. A study by Purtell et al points to the fact that compared to adiposity-matched control subjects, hyperphagia in PWS is not related to a lower postprandial GLP-1 or PYY response and that elevated ghrelin levels in PWS are consistent with increased hunger and are unrelated to insulin levels.68
EATING DISORDERS
Anorexia nervosa
Anorexia nervosa (AN) is the most common cause of weight loss in young women and of admission to child and adolescent services.69 Moreover, anorexia nervosa has the highest mortality of any psychiatric disorder.70 It is characterised by refusal to maintain body weight at or above minimally normal weight, intense fear of gaining weight or becoming fat, and disturbance in the way body weight or shape is experienced. The aetiology of anorexia nervosa is not currently well understood. Recently, some studies have focused on the relationship between hunger regulation and gut-brain peptides; in particular, numerous data show that alterations of central and/or peripheral peptidergic signalling are related to disturbed regulation of feeding and body weight, peptidergic signalling including anorexigenic corticotropic-releasing factor, melanocortin, NPY, and ghrelin.
In terms of ghrelin levels, several studies have reported that individuals with anorexia nervosa have higher levels of fasting plasma ghrelin than normal weight healthy controls. For example, Monteleone et al compared 20 female anorexia nervosa patients with 20 healthy women; it was found that AN patients displayed significantly increased circulating levels of ghrelin compared to healthy patients.71 In another study, Nedvikova et al investigated responses of plasma ghrelin to food intake, meal volume, and meal nutritional value in healthy volunteers and women with anorexia nervosa; it was found that fasting plasma ghrelin was significantly higher in AN patients than in controls and correlated negatively with percentage of body fat in both groups. Ghrelin levels markedly fell after consumption of either a standardised meal or fibre in controls, but not in anorexic women.72 These results indicate that the acute plasma ghrelin response to food intake is impaired in AN patients. The authors suggested that this response could be part of a chronic adaptation to prolonged food restriction which attempts to restore normal feeding conduct by maintaining the drive to eat. Another study by Tolle et al involved assessing ghrelin plasma levels in anorexia nervosa patients before and after re-nutrition and in constitutionally thin subjects with BMIs equivalent to those of AN women but with no abnormal feeding behaviour.73 It was found that morning fasting plasma ghrelin levels were doubled compared with levels in controls and constitutionally thin subjects and that 4- and 24-hour ghrelin levels were increased compared to controls. These results indicate that ghrelin levels are not only dependent on body fat mass but are also affected by nutritional status.
The reduced food intake that characterises anorexia nervosa, despite the chronically increased ghrelin levels, may suggest that this condition represents a state of “ghrelin insensitivity or resistance”. This theory is supported by results that showed that anorexia nervosa patients did not respond to ghrelin administration by increasing appetite and food intake as did healthy controls.74 Ogiso et al comment that although many studies have investigated the association between anorexia nervosa and ghrelin, most studies measured total ghrelin and did not distinguish between acyl ghrelin and desacyl ghrelin.26 This could play a role as to how results are interpreted, as certain studies have found that desacyl ghrelin has opposite effects to acyl ghrelin. For example, Asakawa et al demonstrated that desacyl ghrelin induces a negative energy balance by decreasing food intake and delaying gastric emptying in mice.75 The results of these studies contradict the previously mentioned findings of Heppner et al according to which desacyl ghrelin is an agonist of GHSR leading to increased fat mass and increased glucose-stimulated insulin secretion.22 In a study by Koyama et al, the changes in ghrelin levels (both acyl and desacyl) during early treatment of anorexia nervosa were measured.76 It was found that desacyl ghrelin in anorexia nervosa patients was higher than in control subjects before the therapy, but it decreased with treatment and was significantly lower than in control subjects after 8 weeks of treatment; moreover, the ratio of the acyl ghrelin to total ghrelin increased with 8 weeks’ treatment. These results may suggest that a successful refeeding outcome could involve increasing acyl ghrelin and decreasing desacyl ghrelin. In conclusion, future studies investigating ghrelin levels in anorexia nervosa could measure both acyl and desacyl ghrelin in order to obtain a clearer picture of the role of ghrelin in anorexia nervosa.
It currently remains unknown why ghrelin levels are higher in anorexia nervosa patients than in healthy controls and why people with anorexia nervosa do not respond appropriately to ghrelin. Terashi et al proposed that changes of ghrelin reactive autoantibodies could explain elevated plasma ghrelin in AN.77 Autoantibodies to ghrelin occur naturally, and it is thought that physiologic ghrelin autoantibodies help regulate the ghrelin plasma levels.41 The autoantibodies could alter the feeding-regulatory circuitry and behaviour by changing the signalling of the molecule, ranging from transport to neutralisation.26 In their study, Terashi et al found that patients with anorexia nervosa had significantly lower plasma levels of acyl ghrelin IgG, IgM, and IgA autoantibodies; the levels remained low even after one month of refeeding.77 The authors proposed that the decrease of bioavailable ghrelin autoantibodies may underlie a long-term elevation of plasma ghrelin levels and the resulting phenomenon of ghrelin resistance in malnourished patients with anorexia nervosa.
Differences in ghrelin levels have also been noted in regard to the type of anorexia nervosa. Whereas studies have recorded increased ghrelin levels in restrictive anorexia nervosa, others have reported unchanged ghrelin levels in binge-purge anorexia nervosa.78,79
Further research is needed to fully elucidate the ghrelin effects and the ghrelin level fluctuations in anorexia nervosa. In addition, further investigations are required to determine whether any genes for ghrelin can predispose individuals to anorexia nervosa. It has been hypothesised by Dardennes et al (2007) that ghrelin and AgRP polymorphisms confer susceptibility to anorexia nervosa; the results of their study demonstrated a transmission disequilibrium for the Leu72Met variant of the preproghrelin gene and an excess of transmission of the Gln90Leu72 preproghrelin/obestatin haplotype in patients with anorexia nervosa (bingeing-purging subtype).80 Moreover, Muller et al observed an association of a GOAT gene variant with AN in a study involving 543 German patients with AN and 612 German normal and underweight healthy controls.81 A genetic variation of the ghrelin activator gene MBOAT4 was implicated as a causal factor in anorexia nervosa,81 and the 3056T>C SNP of the ghrelin gene was suggested in another as being related to recovery from restrictive anorexia nervosa.82 However, Kindler et al found no increased occurrence of 3 ghrelin gene polymorphisms in patients with eating disorders compared with in healthy controls.83
Bulimia nervosa
Bulimia nervosa (BN) is characterised by eating in a discrete period of time an amount of food that is definitely larger than most people would eat during a similar period of time and under similar circumstances. In addition, a sense of lack of control over eating during the episode and recurrent inappropriate compensatory behaviour to prevent weight gain characterise the syndrome.84
Ghrelin levels in bulimia nervosa are variable according to the clinical study and various results have been obtained. BMI matched bulimia nervosa patients had significantly higher fasting plasma ghrelin levels compared to healthy volunteers.85,86 However, other studies found no significant differences.71,87 It has been suggested that the differences in the results of these studies could be attributed to the different methods being used, for example radioimmunoassay vs. ELISA, or measuring fasting vs non-fasting ghrelin levels.88 Reduced ghrelin suppression after eating was found in some studies and it has been theorised that this blunted postprandial decrease in ghrelin levels could indicate a reduced satiety response, which could in turn explain the binges.86,89
When comparing bulimia nervosa with anorexia nervosa patients, fasting plasma ghrelin levels were found to be significantly lower in women with binge-eating and purging behaviour compared to restricting type anorexia nervosa patients.90
It is possible that ghrelin gene polymorphisms could be associated with vulnerability for bulimia nervosa. For example, it has been found that the C allele at the 3056T>C SNP (CC and TC genotypes) in intron 3 and the Met allele at the Leu72Met SNP of the ghrelin gene were significantly more frequent in purging-type BN.82 Moreover, a GHSR gene variant that was associated with BN in Japanese patients.91
CACHEXIA
Cachexia is defined as a complex metabolic syndrome associated with underlying illness and characterised by loss of muscle with or without loss of fat mass.92 Cachexia is distinct from starvation, age-related loss of muscle mass, primary depression, malabsorption, and hyperthyroidism and is associated with increased morbidity. It can occur in patients with advanced cancer and/or chronic progressive diseases.93 Malnutrition has been observed in a variety of patients with chronic diseases, including congestive heart failure (CHF), chronic obstructive pulmonary disease (COPD), renal failure, and cancer.94 The inverse relationship between plasma ghrelin and BMI observed in healthy individuals also applies to cachectic patients, for example in underweight patients with chronic obstructive pulmonary disease,95 but there were no significant differences in ghrelin levels between normal subjects and cachectic (CHF, COPD, cancer) patients after matching for BMI.95,96 Some studies, however, have demonstrated elevated levels in cachectic patients with a variety of cancers.97 These elevated ghrelin levels could serve as a compensation for energy loss to maintain homeostasis and as a defence mechanism against starvation. Various factors could contribute to the increased ghrelin levels in certain cancers, and it has been reported that some cancers can express ghrelin.98 Elevated ghrelin levels have also been found in cachectic patients with chronic renal failure,99 but this might be due to increased desacyl ghrelin levels, whereas acyl ghrelin levels are not increased.100 It was suggested that the elevation in total ghrelin is secondary to the fact that desacyl ghrelin is cleared through the kidneys, leading to accumulation in renal insufficiency.41
APPLICATIONS IN CLINICAL PRACTICE
Ghrelin as a pharmacotherapy
Ghrelin administration
In addition to lean and obese healthy volunteers, ghrelin administration studies have involved several patient populations including those with congestive heart failure, several types of cancer, diabetes mellitus, pulmonary disease, anorexia nervosa, end-stage renal disease, Cushing’s syndrome, gastroparesis, polycystic ovary syndrome, hyperthyroidism, hyperparathyroidism, depression, acromegaly, and GH deficiency.101 By reviewing these treatment studies Garin et al concluded that there is strong evidence that ghrelin is an effective appetite stimulant, resulting in increased energy intake but that there is less evidence that ghrelin can cause positive changes in body composition, and almost no evidence of an increase in muscle strength and performance.101
Only a small number of studies have so far investigated the effects of ghrelin administration on patients with anorexia nervosa. The first study to investigate this, by Broglio et al, involved administration of ghrelin to nine women with anorexia nervosa (restricting type) and seven healthy women; the results indicated an impaired GH response to ghrelin in anorexia nervosa. Although food intake was not measured, hunger was mentioned as an adverse event.102 A more recent pilot study by Hotta et al investigated the effects of ghrelin on appetite, energy intake, and nutritional parameters in five patients with restricting-type AN who were fully motivated to gain body weight but could not increase their food intake because of malnutrition-induced gastrointestinal dysfunction. The daily energy intake of the five patients during the pre-treatment period ranged from 825 to 1426 kcal. During ghrelin infusion, four patients showed a statistically significant increase in daily energy intake. Mean increase in daily energy intake during ghrelin infusion was about 20% when compared with the pretreatment period. Analysis of nutrients revealed significant increases in daily intakes of carbohydrate in three patients, fat in one patient, and protein in all patients. The ghrelin administration improved epigastric discomfort and constipation and increased the hunger score.103 The results of these studies indicate that ghrelin could potentially be a new treatment for anorexia nervosa. However, a study by Miljic et al found that ghrelin administration did not significantly affect appetite in 25 young women with anorexia nervosa.74 It has been suggested that these results could be reflecting differences in population, treatment duration, and dose.89
Other studies investigated the effect of ghrelin administration on various cachectic conditions. Nagaya et al administered human synthetic ghrelin intravenously to 10 patients with congestive heart failure for three weeks. It was found that both food intake and body weight were increased; moreover, exercise capacity, muscle wasting, and left ventricular function were improved.104 An open label pilot study by Nagaya et al examined whether ghrelin could improve cachexia and functional capacity in seven cachectic patients with COPD. It was found that a three-week ghrelin treatment resulted in a significant increase in mean body weight, food intake, lean body mass, and peripheral and respiratory muscle strength; moreover, ghrelin attenuated the exaggerated sympathetic nerve activity.105 In a study by Wynne et al, nine peritoneal dialysis patients with mild to moderate malnutrition were administered ghrelin subcutaneously. The results demonstrated a significant increase in the group mean absolute energy intake, compared with placebo; moreover, ghrelin administration resulted in immediate doubling of energy intake when expressed as proportional energy increase for each individual.106 Other studies investigated the effects of administering ghrelin in the setting of cancer cachexia. Although the use of supra-physiological doses of ghrelin was generally required, the effects on appetite have been positive; this suggests that the effects of ghrelin on appetite-stimulating centres are not saturated in the setting of cancer cachexia. A randomised, placebo-controlled, cross-over clinical trial demonstrated appetite stimulation by ghrelin in seven cancer patients with cachexia; the results showed that administration of ghrelin significantly increased food intake and meal appreciation as compared to saline infusion.107 However, there are concerns regarding the use of ghrelin in cancer cachexia; for example, an important concern is that ghrelin may increase growth factors, such as GH and IGF-1, leading to stimulation of tumour growth.108 In addition, the short half-life of ghrelin and the route of delivery via injection are additional limitations of the use of ghrelin as a therapeutic agent.
Apart from exerting anti-cachectic effects by acting on the GHSR1a receptor, Porporato et al proposed that acyl and desacyl ghrelin could act on a common, unidentified receptor to block skeletal muscle atrophy in a GH-independent manner.109 Their results demonstrated that both acyl and desacyl ghrelin inhibited dexamethasone-induced skeletal muscle atrophy and atrogene expression through PI3Kβ-, mTORC2-, and p38-mediated pathways in myotubes. Moreover, it was shown that acyl and desacyl ghrelin induced phosphorylation of Akt in skeletal muscle and impaired fasting-induced atrophy in Ghsr-deficient mice.
A recent double-blind placebo, controlled, cross-over study has indicated that desacyl ghrelin may improve glycaemic control in obese diabetic subjects by decreasing acylated ghrelin level.110 The study involved investigating the effects of continuous overnight infusion of desacyl ghrelin on acylated ghrelin levels and glucose and insulin responses to a standard breakfast meal in eight overweight patients with type 2 diabetes. It was found that, compared with placebo, overnight desacyl administration significantly decreased postprandial glucose levels, both during continuous glucose monitoring and in peak serum glucose levels; moreover, the degree of improvement in glycaemia was correlated with baseline plasma acylated ghrelin levels, which were decreased. The authors of the study suggested that desacyl ghrelin is a good candidate for the development of compounds in the treatment of metabolic disorders and conditions such as type 2 diabetes mellitus and PWS.
The administration of desacyl ghrelin may also have a clinical application. It has previously been demonstrated that desacyl ghrelin can counteract the effects of acyl ghrelin on insulin secretion and glucose metabolism: acyl ghrelin administration was followed by a decrease in insulin levels and an increase in plasma glucose levels; while desacyl ghrelin administration alone had no effects, desacyl ghrelin together with acyl ghrelin diminished the insulin and glucose response to acyl ghrelin.111 Moreover, it has been reported that i.v. desacyl ghrelin administration improves glucose metabolism and inhibits lipolysis in healthy volunteers.112 The results of these studies imply that desacyl ghrelin or desacyl ghrelin analogs might have a pharmacological application in the future treatment of metabolic disorders, diabetes, obesity, and PWS, as they could inhibit or suppress ghrelin.
Ghrelin receptor agonists
Several ghrelin agonists are currently under development for various indications. For example, a recent phase 2a, randomised, double-blind 28-day study of the ghrelin receptor TZP-102 demonstrated a reduction of the symptoms of diabetic gastroparesis.113 Other targets of ghrelin receptor agonists include cancer cachexia, postoperative ileus, and opioid-induced bowel dysfunction.114 Palus et al used a rat model of congestive heart failure to investigate the effects of administering the ghrelin analogues BIM-28125 and BIM-28131; whereas placebo-treated rats gained no fat (but only lean mass), the active compounds induced both fat and lean mass gain.115 In a recent study, Lenk et al also used a rat model of congestive heart failure and showed that the same analogues significantly increased the expression of myostatin in the skeletal muscles of the rats.116 In a human study of healthy volunteers, the oral ghrelin agonist and GH secretagogue RC-1291 produced dose-related increases in body weight with no dose-limiting adverse effects; the authors suggested that RC-1291 could be an effective treatment for cancer-associated cachexia and anorexia.117
Ghrelin receptor antagonists
Various pharmacological tools are being investigated to counteract the effects of ghrelin (Figure 2). The ghrelin receptor antagonists are an example of these tools. A study by Beck et al involved administering the ghrelin receptor antagonist [D-Lys)]-GHRP-6 to fa/fa obese Zucker rats; it was found that the antagonist decreased the food intake of rats.118 Moreover, this antagonist decreased energy intake, rate of gastric emptying, and body weight gain in ob/ob mice.119
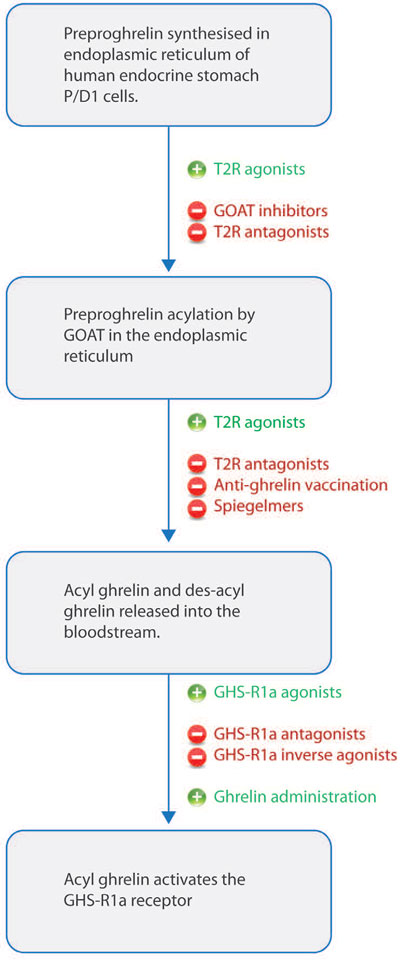
Figure 2. Diagram demonstrating pharmacological tools to enhance or counteract the effects of ghrelin. Synthesis of ghrelin and acylation by GOAT occur in the endoplasmic reticulum. Both acyl and desacyl ghrelin are then secreted into the bloodstream by fusion of secretory vesicles with the plasma membrane. Acyl ghrelin then activates GHSR1a receptors T2R, bitter taste receptor.
Esler et al also showed that the ghrelin antagonist YIL-781 not only reduced fat mass in a mouse model of diet-induced obesity but also directly improved glucose homeostasis by enhancing glucose-stimulated insulin secretion.120 Several compounds have been identified as ghrelin receptor antagonists, including piperidine-substituted quinazolinone derivatives, optimised piperazine-bisamide analogs, and carbohydrazide derivatives. These classes of GHSR1a antagonists could become pharmacological tools for the treatment of obesity, as well as type 2 diabetes and metabolic syndrome. However, long-term human and animal studies are still required to further investigate the beneficial effects of ghrelin antagonists in the context of obesity.
Ghrelin receptor inverse agonists
The ghrelin receptor GHSR1a has a high constitutive activity. It has been proposed that inverse ghrelin receptor agonists could be useful for treating obesity by decreasing this constitutive activity.121 Moreover, it has been hypothesised that the constitutive activity of the receptor may be high between meals, when plasma ghrelin levels are low; hence, it has been suggested that whereas ghrelin receptor antagonists could be used to block the acute preprandial ghrelin signal, inverse ghrelin agonists could block the high constitutive activity.114 The compound [D-Arg1, D-Phe5, D-Trp7,9,Leu11] Substance P was identified as a a low-potency antagonist but a high-potency full inverse agonist on the ghrelin receptor.122 Long-term animal and human studies are necessary to elucidate the beneficial effects of ghrelin receptor inverse agonists in the treatment of obesity.
Ghrelin O-acyltransferase (GOAT) inhibitors
Pharmacological tools have been developed to target the inhibition of GOAT. An example of these is GO-CoA-Tat, which is a peptide-based bisubstrate analog. Barnett et al demonstrated that GO-CoA-Tat potently inhibits GOAT in vitro, in cultured cells, and in mice.123 Moreover, they showed that intraperitoneal administration of GO-CoA-Tat improved glucose tolerance and reduced weight gain in wild-type mice but not in ghrelin-deficient mice. These results indicate that GOAT could be a promising target for the development of anti-obesity and anti-diabetes drugs. In terms of future design of GOAT inhibitors, the results of Yang et al (2008) suggest that GOAT is subjected to end-product inhibition and this inhibition is better achieved with substrates having the octanoyl group attached through an amide linkage rather than the corresponding ester.124
Bitter taste receptors
Bitter taste receptors (T2R) and the gustatory G proteins, α-gustducin (gust) and α-transducin, are expressed in the gut and are involved in the chemosensation of nutrients. Jannsen et al (2011) demonstrated that intragastric administration of T2R-agonists increased food intake during the first 30 min in wild-type but not in gust/ and ghrelin receptor knockout mice.125 Moreover, gavage of T2R-agonists increased plasma octanoyl ghrelin levels in wild-type mice but the effect was partially blunted in gust/ mice. The results of this study suggest that activation of bitter taste receptors stimulates ghrelin secretion, meaning that T2R could be a new pharmacological target for the modulation of ghrelin levels.
Octreotide
Octreotide is a somatostatin receptor type 2 agonist that is used to treat patients with acromegaly or neuroendocrine tumours. Known side-effects of octreotide include decreased insulin secretion, which can lead to impaired glucose tolerance. Norrelund et al (2002) showed that somatostatin infusion lowered ghrelin levels by 70-80% in healthy volunteers.126 In a pilot study by Haqq et al (2003b), short-term octreotide treatment markedly decreased fasting ghrelin concentrations in children with PWS but did not fully ablate the normal meal-related suppression of ghrelin.127 A 56-week prospective, randomised, cross-over trial by De Waele et al (2008) evaluated whether long-acting octreotide decreases acylated and desacyl ghrelin concentrations, body mass, appetite, and compulsive behaviour towards food in adolescents with PWS.128 It was found that octreotide treatment caused a prolonged decrease in ghrelin concentrations in adolescents with PWS but did not improve body mass, appetite or compulsive food-taking behaviour. Based on this evidence, octreotide cannot be currently recommended for the treatment of PWS.128 The authors proposed several possible explanations for these findings. For example, the decrease in circulating ghrelin may be too small to be perceived at the hypothalamic level and translate to clinically-relevant effects, or octreotide may blunt the postprandial decrease in ghrelin concentrations.
Rikkunshito: a ghrelin enhancer
Rikkunshito is a kampo herbal medicine which is widely used in Japan for the treatment of upper gastrointestinal symptoms.129 A study by Matsumura et al (2010) involved administering rikkunshito to healthy volunteers and mice for two weeks and examining the changes in plasma peptide and hormone levels; it was found that rikkunshito increased the plasma acylated ghrelin level in both healthy volunteers and normal mice.130 Apart from stimulating the secretion of ghrelin, rikkunshito can enhance ghrelin’s orexigenic effect by several additional mechanisms; for example, it has been found that rikkunshito increased the reactivity of ghrelin by inhibiting PDE3 activity.131 Examples of indications where rikkunshito use could be beneficial include cisplatin-induced anorexia, anorexia of aging, and stress-related increased or decreased food intake.
Neutralisation of ghrelin
Anti-ghrelin vaccine
Vaccination against ghrelin could be another strategy to block the effects of ghrelin. Active vaccination of mature rats with ghrelin immunoconjugates led to the production of antibodies directed against acylated ghrelin; the antibody production decreased feed efficiency, relative adiposity, and body weight gain.132 In a study involving normal weight mice and mice with diet-induced obesity, vaccination was shown to be effective in decreasing acute food intake and increasing energy expenditure, but there was no change in body weight over the study span.133 In another study, high-affinity anti-acyl ghrelin-specific monoclonal antibodies were generated; although in a 4-week chronic study the antibodies effectively bound to endogenous acyl ghrelin, long-term administration did not affect food intake or body weight gain in a mouse model of diet-induced obesity.134 Moreover, the development of a ghrelin vaccine in Switzerland has been stopped because of obtaining negative phase IIa results in a study with 111 obese subjects; strong antibody responses to ghrelin were elicited leading to reduced hunger, but there was no significant weight loss.135 The results of these studies suggest that although peripheral neutralisation of ghrelin can suppress appetite stimulated by a transient ghrelin surge, compensatory mechanisms contributing to the regulation of energy balance may prevent long-term effects on body weight. Patients that could benefit by an effective anti-ghrelin vaccine could include individuals with PWS. In obesity, most obese individuals have low ghrelin levels and the vaccine may not be effective on its own for this patient group; instead, an anti-ghrelin vaccine could benefit obese patients enrolling in a diet and exercise program as adjuvant therapy for weight loss and bodyweight control.136
Spiegelmers
RNA spiegelmers, meaning “RNA mirrors”, are a novel type of ghrelin blocking reagents. Spiegelmers are oligonucleotides synthesised with unnatural L-enantiomers of ribose in the sugar-phosphate backbone, making them stable in vivo. They have been designed to bind specifically and avidly to acylated ghrelin, thus preventing activation of the ghrelin receptor in vitro.114 The spiegelmer NOX-B11-2 blocked ghrelin mediated activation of GH secretagogue receptor 1a in cell culture and also effectively promoted weight loss in diet-induced obese mice.137 Another spiegelmer, NOX-B11-3, effectively blocked the excitatory effect of ghrelin in the medial arcuate nucleus of rats.138 Moreover, i.v. administration of the spiegelmer NOX-B11 efficiently suppressed ghrelin-induced growth hormone release in rats.139 Further investigation is required to elucidate the beneficial clinical effects of spiegelmers, which could be useful to treat diseases with high ghrelin levels and/or severe obesity, such as PWS.
CONCLUSION
Present evidence suggests that ghrelin plays an important role in obesity and eating disorders, as well as in regulating appetite and energy balance in healthy individuals. The effects of ghrelin can be both homeostatic (under the control of circulating hormones acting primarily on the hypothalamus), as well as non-homeostatic involving control of reward-based eating. In pathological states, ghrelin can be lower than normal, as is seen in obese individuals, or can be higher than normal, as has been reported for PWS, anorexia nervosa, bulimia nervosa, and certain types of cachexia.
In the future, the use of ghrelin as a clinical target seems promising. This could involve the use of pharmacotherapies (such as ghrelin administration, ghrelin agonists/antagonists/inverse agonists, GOAT inhibitors), as well as neutralisation of ghrelin (for example, by vaccines and spiegelmers). Further studies and large clinical trials are necessary to fully determine the role that ghrelin can play in future clinical practice.
REFERENCES
1. Leite-Moreira AF, Rocha-Sousa A, Henriques-Coelho T 2007 Cardiac, skeletal, and smooth muscle regulation by ghrelin. In: L Gerald (ed) Vitam Horm, Academic Press; pp, 207-238.
2. van der Velde M, Delhanty P, van der Eerden B, van der Lely AJ, van Leeuwen J 2007 Ghrelin and Bone. In: L Gerald (ed) Vitam Horm, Academic Press; pp, 239-258.
3. Fuglsang J 2007 Ghrelin in pregnancy and lactation. In: L Gerald (ed) Vitam Horm, Academic Press; pp, 259-284.
4. Tena-Sempere M 2007 Ghrelin and reproduction: ghrelin as novel regulator of the gonadotropic axis. In: L Gerald (ed) Vitam Horm, Academic Press; pp, 285-300.
5. Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, 2013 Intrarenal ghrelin receptors regulate ENaC-dependent sodium reabsorption by a cAMP-dependent pathway. Kidney Int 84: 501-508.
6. Altınova AE, Törüner FB, Aktürk M, et al, 2006 Reduced serum acylated ghrelin levels in patients with hyperthyroidism. Horm Res 65: 295-299.
7. Ates Y, Degertekin B, Erdil A, Yaman H, Dagalp K, 2008 Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig Dis Sci 53: 2215-2221.
8. Freda PU, Reyes CM, Conwell IM, Sundeen RE, Wardlaw SL, 2003 Serum ghrelin levels in acromegaly: effects of surgical and long-acting octreotide therapy. J Clin Endocrinol Metabol 88: 2037-2044.
9. Kadoglou NPE, Lampropoulos S, Kapelouzou A, et al, 2010 Serum levels of apelin and ghrelin in patients with acute coronary syndromes and established coronary artery disease-KOZANI STUDY. Transl Res 155: 238-246.
10. Libè R, Morpurgo PS, Cappiello V, et al, 2005 Ghrelin and adiponectin in patients with Cushing’s disease before and after successful transsphenoidal surgery. Clin Endocrinol (Oxf) 62: 30-36.
11. Monajemzadeh M, Mokhtari S, Motamed F, et al, 2013 Plasma ghrelin levels in children with cystic fibrosis and healthy children. Arch Med Sci 9: 93-97.
12. Nahata M, Muto S, Oridate N, et al, 2012 Impaired ghrelin signaling is associated with gastrointestinal dysmotility in rats with gastroesophageal reflux disease. Am J Physiol Gastrointest Liver Physiol 303: G42-G53.
13. Nweneka C, Prentice A, 2011 Helicobacter pylori infection and circulating ghrelin levels - A systematic review. BMC Gastroenterol 11: 7.
14. Pinsker JE, Ondrasik D, Chan D, et al, 2011 Total and acylated ghrelin levels in children with poor growth. Pediatr Res 69: 517-521.
15. Pöykkö SM, Kellokoski E, Hörkkö S, et al, 2003 Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes 52: 2546-2553.
16. Schöfl C, Horn R, Schill T, et al, 2002 Circulating ghrelin Levels in patients with polycystic ovary syndrome. J Clin Endocrinol Metab 87: 4607-4610.
17. Selimoglu MA, Altinkaynak S, Ertekin V, Akcay F, 2006 Serum ghrelin levels in children with celiac disease. J Clin Gastroenterol 40: 191-194.
18. Yilmaz H, Gerdan V, Kozaci D, et al, 2012 Ghrelin and adipokines as circulating markers of disease activity in patients with Takayasu arteritis. Arthritis Res Ther 14: R272.
19. Kojima M, Hosoda H, Date Y, et al, 1999 Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402: 656-660.
20. Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL, 2008 Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132: 387-396.
21. Hosoda H, Kojima M, Matsuo H, Kangawa K, 2000 Ghrelin and des-acyl ghrelin: Two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem Biophys Res Commun 279: 909-913.
22. Heppner KM, Piechowski CL, Müller A, et al, 2013 Both acyl and des-acyl ghrelin regulate adiposity and glucose metabolism via CNS ghrelin receptors. Diabetes 63: 122-131.
23. Leung P-K, Chow KBS, Lau P-N, et al, 2007 The truncated ghrelin receptor polypeptide (GHS-R1b) acts as a dominant-negative mutant of the ghrelin receptor. Cell Signal 19: 1011-1022.
24. Gnanapavan S, Kola B, Bustin S, et al, 2002 The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab 87: 2988-2988.
25. Lango Allen H, Estrada K, Lettre G, et al, 2010 Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467: 832-838.
26. Ogiso K, Asakawa A, Amitani H, Inui A, 2011 Ghrelin and anorexia nervosa: A psychosomatic perspective. Nutrition 27: 988-993.
27. Amini P, Wadden D, Cahill F, et al, 2012 Serum acylated ghrelin is negatively correlated with the insulin resistance in the CODING study. PLoS ONE 7: e45657.
28. Tong J, Prigeon RL, Davis HW, et al, 2010 Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 59: 2145-2151.
29. Tschop M, Smiley DL, Heiman ML, 2000 Ghrelin induces adiposity in rodents. Nature 407: 908-913.
30. Theander-Carrillo C, Wiedmer P, Cettour-Rose P, et al, 2006 Ghrelin action in the brain controls adipocyte metabolism. J Clin Invest 116: 1983-1993.
31. Cummings DE, Purnell JQ, Frayo RS, et al, 2001 A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50: 1714-1719.
32. Arafat AM, Weickert MO, Adamidou A, et al, 2013 The impact of insulin-independent, glucagon-induced suppression of total ghrelin on satiety in obesity and type 1 diabetes mellitus. J Clin Endocrinol Metab 98: 4133-4142.
33. Abizaid A, Liu Z-W, Andrews ZB, et al, 2006 Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest 116: 3229-3239.
34. Skibicka KP, Shirazi RH, Rabasa-Papio C, et al, 2013 Divergent circuitry underlying food reward and intake effects of ghrelin: Dopaminergic VTA-accumbens projection mediates ghrelin’s effect on food reward but not food intake. Neuropharmacology 73: 274-283.
35. Perelló M, Zigman JM, 2012 The role of ghrelin in reward-based eating. Biol Psychiatry 72: 347-353.
36. Malik S, McGlone F, Bedrossian D, Dagher A, 2008 Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab 7: 400-409.
37. Disse E, Bussier A-L, Veyrat-Durebex C, et al, 2010 Peripheral ghrelin enhances sweet taste food consumption and preference, regardless of its caloric content. Physiol Behav 101: 277-281.
38. Finger B, Dinan T, Cryan J, 2012 Diet-induced obesity blunts the behavioural effects of ghrelin: studies in a mouse-progressive ratio task. Psychopharmacology (Berl) 220: 173-181.
39. Perello M, Sakata I, Birnbaum S, et al, 2010 Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry 67: 880-886.
40. Landgren S, Simms J, Thelle D, et al, 2011 The ghrelin signalling system is involved in the consumption of sweets. PLoS ONE 6: e18170.
41. Hillman JB, Tong J, Tschop M, 2011 Ghrelin biology and its role in weight-related disorders. Discov Med 11: 521.
42. Gil-Campos M, Aguilera CM, Ramirez-Tortosa MC, Cañete R, Gil A, 2010 Fasting and postprandial relationships among plasma leptin, ghrelin, and insulin in prepubertal obese children. Clin Nutr 29: 54-59.
43. Briggs DI, Enriori PJ, Lemus MB, Cowley MA, Andrews ZB, 2010 Diet-induced obesity causes ghrelin resistance in arcuate NPYAgRP neurons. Endocrinology 151: 4745-4755.
44. Ukkola O, Ravussin E, Jacobson P, et al, 2001 Mutations in the preproghrelinghrelin gene associated with obesity in humans. J Clin Endocrinol Metab 86: 3996-3999.
45. Korbonits M, Gueorguiev M, O’Grady E, et al, 2002 A variation in the ghrelin gene increases weight and decreases insulin secretion in tall, obese children. J Clin Endocrinol Metab 87: 4005-4008.
46. Leskelä P, Ukkola O, Vartiainen J, et al, 2009 Fasting plasma total ghrelin concentrations in monozygotic twins discordant for obesity. Metabolism 58: 174-179.
47. Baessler A, Hasinoff MJ, Fischer M, et al, 2005 Genetic linkage and association of the growth hormone secretagogue receptor (ghrelin receptor) gene in human obesity. Diabetes 54: 259-267.
48. Garcia EA, Heude B, Petry CJ, et al, 2008 Ghrelin receptor gene polymorphisms and body size in children and adults. J Clin Endocrinol Metab 93: 4158-4161.
49. Pugliese-Pires PN, Fortin J-P, Arthur T, et al, 2011 Novel inactivating mutations in the GH secretagogue receptor gene in patients with constitutional delay of growth and puberty. Eur J Endocrinol 165: 233-241.
50. Ma X, Lin Y, Lin L, et al, 2012 Ablation of ghrelin receptor in leptin-deficient obob mice has paradoxical effects on glucose homeostasis when compared with ablation of ghrelin in obob mice. Am J Physiol Endocrinol Metab 303: E422-E431.
51. Saeed S, Bech P, Hafeez T, et al, 2013 Changes in levels of peripheral hormones controlling appetite are inconsistent with hyperphagia in leptin-deficient subjects. Endocrine 45: 401-408.
52. Cole SA, Butte NF, Voruganti VS, et al, 2010 Evidence that multiple genetic variants of MC4R play a functional role in the regulation of energy expenditure and appetite in Hispanic children. Am J Clin Nutr 91: 191-199.
53. Karra E, O’Daly OG, Choudhury AI, et al, 2013 A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Invest 123: 3539-3551.
54. Benedict C, Axelsson T, Söderberg S, et al, 2014 Brief communication: The fat mass and obesity-associated gene (FTO) is linked to higher plasma levels of the hunger hormone ghrelin and lower serum levels of the satiety hormone leptin in older adults. Diabetes 63: 3955-3959.
55. Nicholls RD, Knepper JL, 2001 Genome organization, function, and imprinting in Prader-Willi and Angelman Syndromes. Annu Rev Genomics Hum Genet 2: 153-175.
56. Bieth E, Eddiry S, Gaston V, et al, 2014 Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet: doi: 10.1038ejhg.
57. Cummings DE, Weigle DS, Frayo RS, et al, 2002 Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 346: 1623-1630.
58. Haqq AM, Farooqi IS, O’Rahilly S, et al, 2003 Serum ghrelin levels are inversely correlated with body mass index, age, and insulin concentrations in normal children and are markedly increased in Prader-Willi Syndrome. J Clin Endocrinol Metab 88: 174-178.
59. DelParigi A, Tschöp M, Heiman ML, et al, 2002 High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi Syndrome. J Clin Endocrinol Metab 87: 5461-5464.
60. Goldstone AP, Holland AJ, Butler JV, Whittington JE, 2012 Appetite hormones and the transition to hyperphagia in children with Prader-Willi syndrome. Int J Obes 36: 1564-1570.
61. Scerif M, Goldstone AP, Korbonits M, 2011 Ghrelin in obesity and endocrine diseases. Mol Cell Endocrinol 340: 15-25.
62. Choe YH, Song SY, Paik K-H, et al, 2005 Increased density of ghrelin-expressing cells in the gastric fundus and body in Prader-Willi Syndrome. J Clin Endocrinol Metab 90: 5441-5445.
63. Kojima M, Kangawa K, 2005 Ghrelin: Structure and Function. Physiol Rev 85: 495-522.
64. Goldstone AP, 2004 Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab 15: 12-20.
65. Higgins S, Gueorguiev M, Korbonits M, 2007 Ghrelin, the peripheral hunger hormone. Ann Med 39: 116-136.
66. Feigerlová E, Diene G, Conte-Auriol F, et al, 2008 Hyperghrelinemia precedes obesity in Prader-Willi Syndrome. J Clin Endocrinol Metab 93: 2800-2805.
67. Bizzarri C, Rigamonti AE, Luce A, et al, 2010 Children with Prader–Willi syndrome exhibit more evident meal-induced responses in plasma ghrelin and peptide YY levels than obese and lean children. Eur J Endocrinol 162: 499-505.
68. Purtell L, Sze L, Loughnan G, et al, 2011 In adults with Prader–Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-1 levels. Neuropeptides 45: 301-307.
69. Morris J, Twaddle S, 2007 Anorexia nervosa. BMJ 334: 894-898.
70. Hoek HW, 2006 Incidence, prevalence and mortality of anorexia nervosa and other eating disorders. Curr Opin Psychiatry 19: 389-394.
71. Monteleone P, Serritella C, Martiadis V, Scognamiglio P, Maj M, 2008 Plasma obestatin, ghrelin, and ghrelinobestatin ratio are increased in underweight patients with anorexia nervosa but not in symptomatic patients with bulimia nervosa. J Clin Endocrinol Metab 93: 4418-4421.
72. Nedvídková J, Krykorková I, Barták V, et al, 2003 Loss of meal-induced decrease in plasma ghrelin levels in patients with anorexia nervosa. J Clin Endocrinol Metab 88: 1678-1682.
73. Tolle V, Kadem M, Bluet-Pajot M-T, et al, 2003 Balance in ghrelin and leptin plasma levels in anorexia nervosa patients and constitutionally thin women. J Clin Endocrinol Metab 88: 109-116.
74. Miljic D, Pekic S, Djurovic M, et al, 2006 Ghrelin has partial or no effect on appetite, growth hormone, prolactin, and cortisol release in patients with anorexia nervosa. J Clin Endocrinol Metab 91: 1491-1495.
75. Asakawa A, Inui A, Fujimiya M, et al, 2005 Stomach regulates energy balance via acylated ghrelin and desacyl ghrelin. Gut 54: 18-24.
76. Koyama KI, Yasuhara D, Nakahara T, et al, 2010 Changes in acyl ghrelin, des-acyl ghrelin, and ratio of acyl ghrelin to total ghrelin with short-term refeeding in female inpatients with restricting-type anorexia nervosa. Horm Metab Res 42: 595-598.
77. Terashi M, Asakawa A, Harada T, et al, 2011 Ghrelin reactive autoantibodies in restrictive anorexia nervosa. Nutrition 27: 407-413.
78. Sedlackova D, Kopeckova J, Papezova H, et al, 2012 Comparison of a high-carbohydrate and high-protein breakfast effect on plasma ghrelin, obestatin, NPY and PYY levels in women with anorexia and bulimia nervosa. Nutr Metab 9: 52.
79. Germain N, Galusca B, Grouselle D, et al, 2010 Ghrelin and obestatin circadian levels differentiate bingeing-purging from restrictive anorexia nervosa. J Clin Endocrinol Metab 95: 3057-3062.
80. Dardennes RM, Zizzari P, Tolle V, et al, 2007 Family trios analysis of common polymorphisms in the obestatinghrelin, BDNF and AGRP genes in patients with Anorexia nervosa: Association with subtype, body-mass index, severity and age of onset. Psychoneuroendocrinology 32: 106-113.
81. Müller TD, Tschöp MH, Jarick I, et al, 2011 Genetic variation of the ghrelin activator gene ghrelin O-acyltransferase (GOAT) is associated with anorexia nervosa. J Psychiatr Res 45: 706-711.
82. Ando T, Komaki G, Nishimura H, et al, 2010 A ghrelin gene variant may predict crossover rate from restricting-type anorexia nervosa to other phenotypes of eating disorders: a retrospective survival analysis. Psychiatr Genet 20: 153-159.
83. Kindler J, Bailer U, de Zwaan M, et al, 2011 No association of the neuropeptide Y (Leu7Pro) and ghrelin gene (Arg51Gln, Leu72Met, Gln90Leu) single nucleotide polymorphisms with eating disorders. Nord J Psychiatry 65: 203-207.
84. APA 2000 American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Washington.
85. Tanaka M, Naruo T, Muranaga T, et al, 2002 Increased fasting plasma ghrelin levels in patients with bulimia nervosa. Eur J Endocrinol 146: R1-R3.
86. Kojima S, Nakahara T, Nagai N, et al, 2005 Altered ghrelin and peptide YY responses to meals in bulimia nervosa. Clin Endocrinol (Oxf) 62: 74-78.
87. Nakazato M, Hashimoto K, Shiina A, et al, 2004 No changes in serum ghrelin levels in female patients with bulimia nervosa. Prog Neuropsychopharmacol Biol Psychiatry 28: 1181-1184.
88. Atalayer D, Gibson C, Konopacka A, Geliebter A, 2013 Ghrelin and eating disorders. Prog Neuropsychopharmacol Biol Psychiatry 40: 70-82.
89. Cano S, Merkestein M, Skibicka K, Dickson S, Adan RH, 2012 Role of ghrelin in the pathophysiology of eating disorders. CNS Drugs 26: 281-296.
90. Troisi A, Di Lorenzo G, Lega I, et al, 2005 Plasma ghrelin in anorexia, bulimia, and binge-eating disorder: Relations with eating patterns and circulating concentrations of cortisol and thyroid hormones. Neuroendocrinology 81: 259-266.
91. Miyasaka K, Hosoya H, Sekime A, et al, 2006 Association of ghrelin receptor gene polymorphism with bulimia nervosa in a Japanese population. J Neural Transm 113: 1279-1285.
92. Evans WJ, Morley JE, Argilés J, et al, 2008 Cachexia: a new definition. Clin Nutr 27: 793-799.
93. Ashitani J, Matsumoto N, Nakazato M, 2009 Ghrelin and its therapeutic potential for cachectic patients. Peptides 30: 1951-1956.
94. Sharma R, Anker S, 2001 First cachexia symposium, Berlin, Germany, 1st–2nd December, 2000. Eur J Heart Fail 3: 751-754.
95. Itoh T, Nagaya N, Yoshikawa M, et al, 2004 Elevated plasma ghrelin level in underweight patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 170: 879-882.
96. Huang Q, Fan YZ, Ge BJ, Zhu Q, Tu ZY, 2007 Circulating ghrelin in patients with gastric or colorectal cancer. Dig Dis Sci 52: 803-809.
97. Wolf I, Sadetzki S, Kanety H, et al, 2006 Adiponectin, ghrelin, and leptin in cancer cachexia in breast and colon cancer patients. Cancer 106: 966-973.
98. Nikolopoulos D, Theocharis S, Kouraklis G, 2010 Ghrelin: A potential therapeutic target for cancer. Regul Pept 163: 7-17.
99. Aygen B, Dogukan A, Dursun F, et al, 2009 Ghrelin and obestatin levels in end-stage renal disease. J Int Med Res 37: 757-765.
100. Yoshimoto A, Mori K, Sugawara A, et al, 2002 Plasma ghrelin and desacyl ghrelin concentrations in renal failure. J Am Soc Nephrol 13: 2748-2752.
101. Garin MC, Burns CM, Kaul S, Cappola AR, 2013 The human experience with ghrelin administration. J Clin Endocrinol Metab 98: 1826-1837.
102. Broglio F, Gianotti L, Destefanis S, et al, 2004 The endocrine response to acute ghrelin administration is blunted in patients with anorexia nervosa, a ghrelin hypersecretory state. Clin Endocrinol (Oxf) 60: 592-599.
103. Hotta M, Ohwada R, Akamizu T, et al, 2009 Ghrelin increases hunger and food intake in patients with restricting-type anorexia nervosa: A pilot study. Endocr J 56: 1119-1128.
104. Nagaya N, Moriya J, Yasumura Y, et al, 2004 Effects of ghrelin administration on left ventricular function, exercise capacity, and muscle wasting in patients with chronic heart failure. Circulation 110: 3674-3679.
105. Nagaya N, Itoh T, Murakami S, et al, 2005 Treatment of cachexia with ghrelin in patients with copd. Chest 128: 1187-1193.
106. Wynne K, Giannitsopoulou K, Small CJ, et al, 2005 Subcutaneous ghrelin enhances acute food intake in malnourished patients who receive maintenance peritoneal dialysis: A randomized, placebo-controlled trial. J Am Soc Nephrol 16: 2111-2118.
107. Neary NM, Small CJ, Wren AM, et al, 2004 Ghrelin increases energy intake in cancer patients with impaired appetite: acute, randomized, placebo-controlled trial. J Clin Endocrinol Metab 89: 2832-2836.
108. Akamizu T, Kangawa K, 2011 Therapeutic applications of ghrelin to cachexia utilizing its appetite-stimulating effect. Peptides 32: 2295-2300.
109. Porporato PE, Filigheddu N, Reano S, et al, 2013 Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 123: 611-622.
110. Özcan B, Neggers SJ, Miller AR, et al, 2013 Does des-acyl ghrelin improve glycemic control in obese diabetic subjects by decreasing acylated ghrelin levels? Eur J Endocrinol 170: 799-807.
111. Broglio F, Gottero C, Prodam F, et al, 2004 Non-acylated ghrelin counteracts the metabolic but not the neuroendocrine response to acylated ghrelin in humans. J Clin Endocrinol Metab 89: 3062-3065.
112. Benso A, St-Pierre DH, Prodam F, et al, 2012 Metabolic effects of overnight continuous infusion of unacylated ghrelin in humans. Eur J Endocrinol 166: 911-916.
113. Ejskjaer N, Wo JM, Esfandyari T, et al, 2013 A phase 2a, randomized, double-blind 28-day study of TZP-102 a ghrelin receptor agonist for diabetic gastroparesis. Neurogastroenterol Motil 25: e140-e150.
114. Depoortere I, 2009 Targeting the ghrelin receptor to regulate food intake. Regul Pept 156: 13-23.
115. Palus S, Schur R, Akashi YJ, et al, 2011 Ghrelin and its analogues, BIM-28131 and BIM-28125, improve body weight and regulate the expression of MuRF-1 and MAFbx in a rat heart failure model. PLoS ONE 6: e26865.
116. Lenk K, Palus S, Schur R, et al, 2013 Effect of ghrelin and its analogues, BIM-28131 and BIM-28125, on the expression of myostatin in a rat heart failure model. J Cachexia Sarcopenia Muscle 4: 63-69.
117. Garcia JM, Polvino WJ, 2007 Effect on body weight and safety of RC-1291, a novel, orally available ghrelin mimetic and growth hormone secretagogue: Results of a phase I, randomized, placebo-controlled, multiple-dose study in healthy volunteers. Oncologist 12: 594-600.
118. Beck B, Richy S, Stricker-Krongrad A, 2004 Feeding response to ghrelin agonist and antagonist in lean and obese Zucker rats. Life Sci 76: 473-478.
119. Asakawa A, Inui A, Kaga T, et al, 2003 Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut 52: 947-952.
120. Esler WP, Rudolph J, Claus TH, et al, 2007 Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology 148: 5175-5185.
121. Delporte C, 2012 Recent advances in potential clinical application of ghrelin in obesity. J Obes 2012: doi: 10.11552012535624.
122. Holst B, Lang M, Brandt E, et al, 2006 Ghrelin receptor inverse agonists: identification of an active peptide core and its interaction epitopes on the receptor. Mol Pharmacol 70: 936-946.
123. Barnett BP, Hwang Y, Taylor MS, et al, 2010 Glucose and weight control in mice with a designed ghrelin O-acyltransferase inhibitor. Science 330: 1689-1692.
124. Yang TJ, Zhao TJ, Goldstein JL, Brown MS, 2008 Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proc Natl Acad Sci U S A 105: 10750-10755.
125. Janssen S, Laermans J, Verhulst PJ, et al, 2011 Bitter taste receptors and α-gustducin regulate the secretion of ghrelin with functional effects on food intake and gastric emptying. Proc Natl Acad Sci U S A 108: 2094-2099.
126. Nørrelund H, Hansen TK, Ørskov H, et al, 2002 Ghrelin immunoreactivity in human plasma is suppressed by somatostatin. Clin Endocrinol (Oxf) 57: 539-546.
127. Haqq AM, Stadler DD, Rosenfeld RG, et al, 2003 Circulating ghrelin levels are suppressed by meals and octreotide therapy in children with Prader-Willi Syndrome. J Clin Endocrinol Metab 88: 3573-3576.
128. De Waele K, Ishkanian SL, Bogarin R, et al, 2008 Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader–Willi syndrome. Eur J Endocrinol 159: 381-388.
129. Takeda H, Muto S, Nakagawa K, et al 2012 Chapter Twenty-One - Rikkunshito as a ghrelin enhancer. In: K Masayasu and K Kenji (eds) Methods in Enzymology, Academic Press; pp, 333-351.
130. Matsumura T, Arai M, Yonemitsu Y, et al, 2010 The traditional Japanese medicine Rikkunshito increases the plasma level of ghrelin in humans and mice. J Gastroenterol 45: 300-307.
131. Takeda H, Muto S, Hattori T, et al, 2010 Rikkunshito ameliorates the aging-associated decrease in ghrelin receptor reactivity via phosphodiesterase III inhibition. Endocrinology 151: 244-252.
132. Zorrilla EP, Iwasaki S, Moss JA, et al, 2006 Vaccination against weight gain. Proc Natl Acad Sci U S A 103: 13226-13231.
133. Andrade S, Carreira MC, Ribeiro A, et al, 2011 Development of an anti-ghrelin vaccine for obesity treatment. Endocr Rev 32: P2-305.
134. Lu SC, Xu J, Chinookoswong N, et al, 2009 An acyl-ghrelin-specific neutralizing antibody inhibits the acute ghrelin-mediated orexigenic effects in mice. Mol Pharmacol 75: 901-907.
135. Cytos Biotechnology 2006. Phase IIIa clinical trial with obese individuals shows no effect of CYT009-GhrQb on weight loss. http:www.cytos.comuploadsnewsid180Cytos Press E 061107.pdf Accessed 22nd September 2014.
136. Monteiro MP, 2011 Anti-ghrelin vaccine for obesity: a feasible alternative to dieting? Expert Rev Vaccines 10: 1363-1365.
137. Shearman LP, Wang SP, Helmling S, et al, 2006 Ghrelin neutralization by a Ribonucleic Acid-SPM ameliorates obesity in diet-induced obese mice. Endocrinology 147: 1517-1526.
138. Becskei C, Bilik KU, Klussmann S, et al, 2008 The anti-ghrelin spiegelmer NOX-B11-3 blocks ghrelin- but not fasting-induced neuronal activation in the hypothalamic arcuate nucleus. J Neuroendocrinol 20: 85-92.
139. Helmling S, Maasch C, Eulberg D, et al, 2004 Inhibition of ghrelin action in vitro and in vivo by an RNA-Spiegelmer. Proc Natl Acad Sci U S A 101: 13174-13179.
Address for correspondence:
Márta Korbonits, MD, PhD, Professor of Endocrinology and Metabolism, Centre for Endocrinology, William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK Tel.: +44 20 7882 8284, e-mail: m.korbonits@qmul.ac.uk
Received: 20-09-14, Accepted: 24-09-14