Division of Endocrinology and Metabolism, Bone Metabolic Unit, AHEPA University Hospital, Thessaloniki, Greece
Postprandial variation of bone turnover markers and the closed relationship between bone remodeling and nutrient supply has been extensively studied in the past few years, but the underlying pathophysiologic mechanisms remain largely unknown. Recent studies have shown that the acute regulation of bone turnover induced by feeding is probably mediated by gastrointestinal (GI) peptides. The greater response of bone remodeling during oral versus intravenous glucose administration and the inhibition of this response after administration of octreotide, that inhibits the release of GI peptides, further support the existence of a gut-bone axis. Glucose-dependent insulinotropic peptide and glucagon-like peptides-1 and -2 are released from K and L cells of the gastrointestinal tract, respectively, and are considered the main mediators of the postprandial response of bone turnover. In this review we outline the most recent evidence that demonstrates the role of incretins in nutrient-dependent regulation of bone metabolism. Further elucidation of the underlying mechanisms can be exploited therapeutically in the future.
Bone Remodeling, GIP, GLP-1, GLP-2, Incretins
INTRODUCTION
Bone is a dynamic tissue continuously remodeled throughout life in order to adapt to the mechanical stresses and needs of the developing human skeleton. Preservation of bone mass and structure is of critical importance and is supported by the tight coordination of osteoblastic bone formation and osteoclastic bone resorption. This dynamic process of bone remodeling during adult life reflects on the circulating concentrations of bone matrix proteins and products of collagen metabolism.
Osteoclastic bone resorption can be easily assessed by measuring plasma levels of collagen fragments derived from the degradation of the C- and N-terminal telopeptide region of collagen type I (β-CTX and NTX, respectively).1 Bone formation, on the other hand, can be assessed in serum either by measuring the concentrations of the non-collagenous bone matrix protein, osteocalcin and the procollagen type I N-terminal propeptide (PINP), which are produced by osteoblasts and are released into the circulation during bone formation,2 or by the measurement of the bone fraction of the enzyme alkaline phosphatase that is being cleaved from the cell membrane and enters the circulation during mineralization. The ability to have a direct and reliable estimation of osteoblast and osteoclast activity by the assessment of bone markers in plasma provides valuable information about bone remodeling in health and disease.
THE CIRCADIAN RHYTHM OF BONE TURNOVER
Earlier studies had demonstrated that bone markers undergo circadian periodicity with high values during the night3-7 and lower levels during daytime, with greater amplitude for bone resorption compared to bone formation markers. Researchers have attempted to explain this phenomenon by focusing their studies on hormones that also exhibit diurnal variation such as cortisol and parathyroid hormone. However, it has been shown that the abolition of the morning peak of cortisol following the administration of metyrapone had no effect on the circadian rhythm of bone resorption,8 and similar results were observed after abolition of the circadian rhythm of serum PTH by continuous infusion of calcium.9 Studies also concentrated on the pineal hormone melatonin, whose plasma concentration is 10-50 times higher during the night and could potentially be associated with nocturnal increase in bone resorption.10,11 However, research on blind patients, who lack the circadian variation of melatonin, showed no change of the pattern of osteoclastic activity.12
POSTPRANDIAL VARIATION OF BONE TURNOVER
The turning point for the research on the periodic variation of bone turnover was the observation that bone resorption was significantly impaired during fasting.13 It was proposed that food intake rather than the circadian rhythm was the main cause of changes in markers of bone resorption during a 24-hour period. The postprandial effect on bone resorption appears to be independent of gender, age and menopausal status.12,14 In contrast to the markers of bone resorption, markers of bone formation do not show significant changes following a meal, a phenomenon explained by the uncoupling of the two processes postprandially.15 It has been postulated that the nocturnal increase of bone resorption is a result of reduced supply of nutrients and organic elements that are essential for maintaining calcium homeostasis and cell proliferation processes, such as hematopoiesis and epithelial regeneration. To cope with this, the body mobilizes the reservoir of nutrients and organic components of the skeleton by activating bone resorption. In this manner, the skeleton supplies the body with the elements necessary for survival when there is no supplementation from the environment. On the other hand, postprandial availability of essential nutrients eliminates the need to use the stored elements, resulting in an instant reduction of bone resorption. Indeed, the effect of a meal is of rapid onset and short duration suggesting a non-transcriptional cascade effect.
Despite the significance of the nutrient-dependent regulation of bone turnover in the general energy-homeostasis of the body and the functional integrity of the bone tissue, less is known about the postprandial adaptation of the skeleton in diseases that affect bone metabolism. Two recent studies have investigated this phenomenon in patients with type 2 diabetes mellitus,16 thyroid diseases and beta-thalassemia major,17 all conditions that are very often complicated with low bone mass and increased fracture risk. It has been shown that the suppression of postprandial bone resorption is attenuated in patients with overt diabetes but not in those with impaired glucose tolerance (IGT), suggesting an additional contributing factor in the deterioration of bone quality and bone mass seen in diabetes.16 We have also shown that in patients with hyperthyroidism and beta-thalassemia major, despite the high bone turnover state observed at baseline, the postprandial reduction of bone resorption remains unaltered, while in hypothyroid patients the postprandial suppression of bone resorption is significantly augmented, regardless of the severity of the disease.17
THE QUEST FOR THE MEDIATOR(S) OF NUTRIENT-DEPENDENT REGULATION OF BONE REMODELING
Several hormones are secreted in response to food entering the gastrointestinal system such as insulin, amylin, glucagon, leptin and GH or GH secretagogues, and thus were thoroughly investigated for their potential effect on postprandial reduction of bone resorption.18 However, none of them appeared to be significantly involved in the acute postprandial reduction of bone resorption, which occurs already in the first two hours postprandially and is fully reversible afterwards.
Further research led to the hypothesis that postprandial reduction of bone resorption is regulated by signals from the gastrointestinal tract. The greater response of bone remodeling during oral glucose administration versus intravenous glucose administration15 and the inhibition of this response after administration of octreotide that inhibits the release of gastrointestinal peptides19 further supported this notion, suggesting the existence of a gut-bone axis.
INCRETIN HORMONES AND BONE METABOLISM
Glucose-dependent insulinotropic polypeptide
Glucose-dependent insulinotropic peptide (GIP) is a 42 amino acid peptide synthesized and secreted by K cells of the duodenum in response to nutrients, especially fat.20,21 Since its initial isolation from porcine intestine in 1970 on the basis of its ability to inhibit gastric acid secretion,22 numerous subsequent studies have demonstrated a broader role of GIP in multiple metabolic processes of the body.23-27 Apart from the well-documented role of GIP in the stimulation of insulin secretion from pancreatic beta-cells via glucose-dependent mechanisms (incretin effect),24,25 a survey by Usdin et al 28 identified GIP receptors (GIPR) in a wide range of tissues, such as the adrenal cortex, pituitary gland, heart, brain, adipose tissue, bone tissue and endothelial cells in several vascular beds. In accordance with the wide GIPR expression, GIP has been reported to regulate lipid metabolism27 as well as enteric and splanchnic blood flow.23,26
The main determinant of GIP metabolism is the enzyme dipeptidyl-peptidase-4. GIP is rapidly metabolized after its secretion by this specific dipeptidyl-peptidase of the gut, having a half-life of 2-4 minutes.29 This enzyme, which is also responsible for the metabolism of other gastrointestinal peptides such as GLP1 and GLP-2 to inactive-truncated products, is a 766 amino acid peptidase showing a wide tissue distribution. It acts preferentially on substrates containing the amino acids proline or alanine at position 2 of the N-terminal and occurs in two isoforms, a transmembrane and a soluble one that circulates in plasma. It is mainly the transmembrane isoform that is considered to exert its enzymatic activity.
GIP and bone remodeling
Several recent studies have demonstrated an important role of GIP on bone metabolism.30-36 In vivo studies with genetically altered mice models with overexpression of GIP or complete absence (knockout) of the GIP receptor have shown significant alterations in the bone phenotype of adult animals.30,31
In the first study, the generation of transgenic mice overexpressing GIP under the control of the metallothionein promoter (Tg+) resulted in higher mean GIP levels in Tg+ mice compared to normal controls.30 These researchers demonstrated that Tg+ animals also had a significant increase in markers of bone formation, a decrease in markers of bone resorption and a significant increase in bone mass as assessed by densitometry and histomorphometry. Based on this evidence, it has been proposed that excess signaling through the GIP receptor in bone can uncouple bone formation from bone resorption in favor of the former and lead to significant gains in bone mass.30
On the other hand, GIPR knockout mice (GIPR-/-) exhibited decreased bone size, and bone mass, deterioration of bone microarchitecture and altered biomechanical properties.31 The effect of the absence of GIP signaling in bone tissue was site-specific and, most interestingly, over time losses in bone mass were partly restored, showing that compensatory mechanisms were developed to ameliorate the negative impact of the absence of GIP signaling on bone. In addition GIPR-/- mice had earlier age-related changes in bone mass and fat percentage compared to wild-type mice, suggesting a more general role for GIP in the whole body composition of the developing organism.31 These in vivo data indicated that GIP has a significant anabolic effect on bone mass and bone quality.
At the cellular level, the presence of GIPR has been identified in a variety of cell-residents in the bone microenvironment, such as osteoblasts, osteoclasts, osteocytes, chondrocytes and bone marrow pluripotent mesenchymal cells.32-36 Moreover, GIP receptors in bone cells show a similar degree of affinity with their respective receptors in beta cells of pancreatic islets, demonstrating the functional importance of these receptors.37
In osteoblasts, activation of GIPR after GIP binding increases the expression of type 1 collagen and alkaline phosphatase activity.32,33 Moreover, the addition of GIP in cultured osteoblast precursors promotes their differentiation, increases their proliferation and also displays anti-apoptotic activity in multipotent mesenchymal cells in bone marrow.34,35 Although osteoclasts were also found positive for GIPR expression,36 studies investigating a direct role of GIP on osteoclast function presented controversial results.34,36
In the study by Zhong et al,36 GIP exerted a direct action on osteoclasts through GIPR and inhibited PTH-induced bone resorption, while in the fetal long bone resorption assay GIP by itself had no effect on bone resorption. What has been clearly shown in this study was that in mature osteoclasts (i.e. osteoclasts treated with MCSF and RANKL), GIP inhibits active osteoclast resorptive activity, as assessed by osteoclast pit formation assay, and decreases the expression of osteoclast-differentiation markers, such as the enzymes TRAP and cathepsin K and the M-CSF-receptor (c-fms). Tsukiyama et al,34 on the other hand, performed bone histomorphometric analysis of GIP receptor knockout mice and reported that, in the absence of GIP, the bones of these mice show a marked increase in the number of mature osteoclasts and a decrease in osteoblastic bone formation. Using dentin slices, these researchers demonstrated that GIP does not inhibit osteoclastic pit formation. The results of these two studies,34,36 although possibly appearing to be in conflict with each other, actually support the pathophysiological role of GIP as has been proposed by in vivo studies. GIP is secreted only after a meal, and the arrival of nutrients in the bone would be the signal to suppress active bone resorption that occurs during fasting. Therefore, it seems plausible that GIP exerts, either directly or indirectly, a differential effect on osteoclast differentiation and function based on the energy load and the current needs of the bone tissue as an integrated unit (Figure 1).
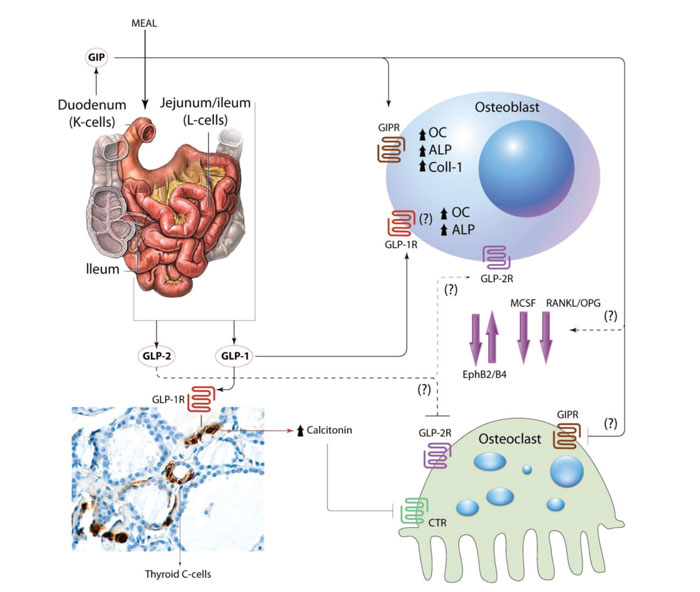
Figure 1. The role of incretins in bone metabolism. Food intake induces secretion of GIP and GLP-1, -2 from K and L cells, respectively. GIP binds to GIPR in osteoblasts to up-regulate production of osteocalcin (OC), alkaline phosphatase (ALP) and collagen type 1 (Coll-1). In osteoclasts it decreases bone resorption. GLP-1 increases production of OC and ALP in osteoblasts via a GLP-1R that may be different from the one identified in pancreatic tissue. In osteoclasts, it seems to exert its action indirectly through up-regulation of calcitonin production from thyroid C-cells. The mechanisms underlying GLP-2 effect on bone remain unknown. The role of GIP and GLP-1 in the crosstalk between osteoblasts and osteoclasts through the expression of Macrophage colony-stimulating factor (MCSF), Receptor activator of nuclear factor kappa-B ligand (RANKL) and Ephrins has not been clarified yet. CTR: calcitonin receptor.
Although the expression of the membrane-bound form of DPP-4 was demonstrated in primary human osteoblasts but not in transformed cell lines (MG63 and SaOs-2),38 the potential contribution of this enzyme in the less pronounced expression of GIPR and GIP effect in primary human osteoblasts32 has not yet been clarified.
Collectively, these data suggest that GIP may be one of the major hormones linking nutrient ingestion to bone formation.
Studies in mice have shown that antagonism of the GIP receptor with proline-3 gastric inhibitory polypeptide ((Pro(3))GIP), which is resistant to DPP-4, and its longer-acting form (Pro(3))GIP mini-polyethylene glycol ((Pro(3))GIP[mPEG]), can reverse or even prevent many of the metabolic abnormalities associated with diet-induced obesity-diabetes (diabesity).39,40 Similar results were obtained from GIP analogues resistant to DPP-4, such as the [D-Ala(2)]GIP in Vancouver Diabetic Fatty (VDF) Zucker rats.41 GIP signaling presents a promising therapeutic target for the management of type 2 diabetes and obesity and thus further investigation of GIP’s linking role between nutrient ingestion and bone metabolism is of critical importance.
GLUCAGON-LIKE PEPTIDE-1 (GLP-1)
GLP-1 is produced by the L-cells of the gut after food intake in two biologically active forms: amide-GLP-1 (7-36) and GLP-1 (7-37), and, as with GIP, is rapidly degraded by DPP-4. GLP-1 promotes glucose-dependent insulin secretion42 and increases insulin synthesis, inhibits glucagon secretion and gastric emptying and displays anorectic action in the central nervous system.43 Moreover, it has been shown that it exerts proliferative and anti-apoptotic actions in the islet pancreatic beta cells and promotes angiogenesis. Finally, there are studies suggesting a possible protective effect in the cardiovascular and central nervous systems.44
GLP-1 AND BONE METABOLISM
The first clue to a potential role of GLP-1 in bone metabolism came from a study by Yamada et al in GLP-1 receptor knockout mice (GLP-1R-/-).45 In this model, genetic disruption of GLP-1 receptor signaling led to cortical osteopenia and bone fragility, as assessed by bone densitometry, as well as increased osteoclastic numbers and bone resorption activity, assessed by histomorphometry.45 However, GLP-1 was not found to exert a direct effect on osteoclasts or osteoblasts. Investigating further the underlying molecular mechanisms for the bone phenotype observed in GLP-1R-/- mice, researchers suggested that thyroid-produced calcitonin could, at least partly, mediate the effect of endogenous GLP-1 receptor signaling in bone.45 Providing proof of this, GLP-1R-/- mice exhibited higher levels of urinary deoxypyridinoline, a marker of bone resorption, and reduced levels of calcitonin mRNA transcripts in the thyroid, while calcitonin treatment in these mice suppressed increased deoxypyridinoline. Moreover, GLP-1R have been identified in thyroid-C cells46 and administration of exendin-4 (Ex-4), a DPP-4 resistant and long-acting GLP-1 receptor agonist, increased calcitonin gene expression in the thyroid of wild-type mice,47 suggesting that lack of GLP-1 receptor signaling increases osteoclastic bone resorption through reduced thyroid calcitonin expression.
More recent in vivo studies have shown that GLP-1 can also have an anabolic effect on bone independent of its insulinotropic action.48-50 In studies with streptozotocin-induced type 2 diabetic (T2D) and fructose-induced insulin-resistant (IR) rats, administration of GLP-1 for 3 consecutive days reversed hyperglycemia and significantly improved the trabecular bone microarchitecture (increased anisotropy) and mechanical properties and up-regulated the expression of bone formation markers, in an insulin and PTH-independent manner.48 Similar results were obtained by administration of exendin-4, which was shown to promote bone formation in diabetic and insulin-resistant rats by interacting with the Wnt signaling pathway.49 The same research group evaluated further the osteogenic properties of GLP-1 and exendin-4 in a hyperlipidic (HL) and hypercaloric rat model which is used for the study of the fat-bone axis.50 The unique characteristic of this model is that it demonstrates the metabolic consequences of obesity without changes in body weight and therefore it is used to study the deleterious effect of hyperlipidemia on bone metabolism without protective involvement of weight gain and the interference of an increased mechanical loading. In this study,50 HL rats demonstrated decreased BMD and bone mineral content (BMC) and reduced OPG/RANKL ratio in the tibia, which were restored after administration of GLP-1 or Ex-4, with Ex-4 showing higher efficiency compared to GLP-1.
At the cellular level, the mechanism by which GLP-1 exerts its osteogenic effect remains largely unknown. The G protein-coupled GLP-1 R is expressed on osteoblastic precursor cells, but not on mature osteoblasts,33 suggesting that GLP-1-regulated osteoblast activity depends on the osteoblastic differentiation stage. In a recent study, GLP-1 acting through the pancreatic GLP-1 receptor was shown to prevent the differentiation of human bone marrow stromal cells into adipocytes.51 However, researchers did not examine the osteogenic differentiation of these cells after administration of GLP-1. In another study, GLP-1 directly up-regulated osteocalcin expression and decreased expression of Runx-2 in the well characterized later stage osteoblastic cell line (MC3T3-E1 cells), acting directly through a different receptor compared to the one that has been described in pancreatic cells.52 In this study, GLP-1 was shown to act directly on osteoblasts via a GPI/IPG (glycosyl-phosphatidyl-inositols generating short-lived inositol-phosphoglycans) receptor that activates the kinase pathways mitogen-activated protein kinase (MAPK) and inositol phosphate 3 kinase (PI3K).
This observation is in line with data from GLP-1 R signaling in other tissues, such as liver and muscle53 where GLP-1 regulation of glucose homeostasis was not mediated through stimulation of intracellular cAMP, as is in beta pancreatic cells, but through a rapid hydrolysis of glycosylphosphatidylinositols (GIPs), generating inositolphosphoglycans (IPGs) and PI3K and MAPK activities.
However, the aforementioned receptor type in osteoblasts did not bind Ex-4 but only GLP-1.52
GLP-1 analogues resistant to metabolism by dipeptidyl-peptidase-4 (DPP-4), such as exenatide and liraglutide, are being introduced into clinical practice and have proved highly efficacious in the control of hyperglycemia in diabetic patients. Moreover, ongoing Phase III clinical trials are currently investigating the efficacy of long-acting GLP-1 R analogs in the treatment of obesity.
Bone mineral density and the bone formation marker serum alkaline phosphatase were not significantly affected after 44 weeks treatment with exenatide, in comparison to the long-acting insulin glargine, in type 2 diabetic subjects.54 On the other hand, long-term exposure of type 2 diabetic patients to exenatide did not increase fracture risk despite the significant weight loss,55 suggesting that a positive effect of exenatide in bone metabolism could compensate for the decrease in bone mass that would otherwise be expected.56 More studies are warranted in order to clarify the potential role of GLP-1 analogues in the disturbed bone metabolism that is commonly seen in patients with type 2 diabetes.
THE GLUCAGON-LIKE PEPTIDE-2 (GLP-2)
The peptide GLP-2, as GLP-1, is produced by the L-cells of the gut and derives from post-translational modification of the common precursor molecule, the pro-glucagon. The most well studied action of GLP-2 is the decrease in the apoptotic rate of the intestinal epithelial cells. Moreover, it regulates intestinal transport of glucose, food intake, gastric secretion and gastric emptying and improves the function of the intestinal barrier.57 Recent studies have shown that GLP-2 also has a positive effect on bone metabolism.
GLP-2 AND BONE METABOLISM
Administration of GLP-2 in a single dose significantly reduced levels of β-CTX in postmenopausal women.58 Similarly deoxy-pyridinoline (DPD) levels, which also reflect the degree of bone resorption, decreased significantly after the administration of 800 mg GLP-2.18 The inhibitory action of GLP-2 in bone resorption was confirmed in another study where the GLP-2 was administered to postmenopausal women for 14 days at a dosage of 1.6 or 3.2 mg as a subcutaneous injection.59 In this study the levels of markers of bone resorption, serum β-CTX and urine DPD decreased significantly after a period of 14 days, while no significant change in markers of bone formation, osteocalcin and P1NP was observed.59 Finally, in a longer-term clinical study, GLP-2 was administered at different doses (0.4, 1.6 and 3.2 mgr) once daily, in the evening for 4 months in postmenopausal women.60 In this study, administration of GLP-2 decreased the nocturnal increase in markers of bone resorption without causing significant changes in markers of bone formation, and induced significant dose-related gains in bone mineral density of the hip at the end of the study. Prolonged exposure to GLP-2, even with lower concentrations, appeared more effective than high concentrations obtained by iv administration, with respect to β-CTX suppression, suggesting that GLP-2 agonists for osteoporosis treatment should be long-acting for best efficacy.61
Despite the large amount of data on humans, the molecular mechanism underlying GLP-2 regulation of bone remodeling is far from being elucidated. At the cellular level, GLP-2 receptors have been identified in osteoclasts and early osteoblasts but data are still inconclusive33,58 (Figure 1). GLP-2 analogues are currently under intensive research for their potential use in the therapeutic management of patients with short bowel syndrome and inflammatory bowel diseases. Teduglutide, a DPP-4 resistant and long-acting analogue of GLP-2, has been shown to improve intestinal rehabilitation by promoting mucosal growth and possibly by restoring gastric emptying and secretion, thereby reducing intestinal losses and promoting intestinal absorption in phase II clinical trials in patients with short bowel syndrome and intestinal failure.62 With the prospect of introducing into clinical practice GLP-2 analogues resistant to DPP-4, further investigation of GLP-2 mechanisms of action in bone is warranted.
CONCLUDING REMARKS
In the last few years, a growing number of studies have reported positive effects of the gastrointestinal peptides, GLP-1, GIP, and GLP-2 in bone and a new concept for a nutrient-dependent regulation of bone remodeling has been developed. The mechanisms through which feeding regulates bone turnover are much more complex than was originally thought. Current and future use of DPP-4 resistant analogues of the GI peptides as therapeutic targets for different kinds of diseases such as diabetes, bowel disease and obesity may facilitate a greater understanding of the molecular mechanisms that regulate the crosstalk between the gut and bone tissue, an urgent need and an objective that is currently under intensive research.
Drugs that are already widely used for the treatment of diabetes, i.e. DPP-4 inhibitors (DPP-4i), would be expected, since they increase bioavailability of GIP, GLP-1 and GLP-2, to exert a protective effect in bone. This possibility has been evaluated in small-sized clinical trials. In line with the molecular mechanism of action of DPP-4i, in a recent meta-analysis that included 28 clinical trials of a duration of at least 24 weeks, DPP-4i, compared with placebo or other treatments, were associated with reduced risk of fractures, data that remained robust even after the exclusion of comparisons with thiazolidinediones or sulfonylureas.63
Data of GLP-1 analogues on fracture risk in patients with diabetes type 2 are scarce and inconclusive and long-term studies with measurement of bone markers, bone density and clinical fractures rates are required. Despite intensive research on the various body systems, in many cases there is uncertainty regarding the pathways by which the incretins mediate their pleiotropic effects. A rudimentary understanding of the underlying cellular mechanisms involved is urgently needed to shed light on this complex and fascinating concept of the gut-bone axis.REFERENCES
1.Rosenquist C, Fledelius C, Christgau S, et al, 1998 Serum CrossLaps One Step ELISA. First application of monoclonal antibodies for measurement in serum of bone-related degradation products from C-terminal telopeptides of type I collagen. Clin Chem 44: 2281-2289.
2.Rosenquist C, Qvist P, Bjarnason N, Christiansen C, 1995 Measurement of a more stable region of osteocalcin in serum by ELISA with two monoclonal antibodies. Clin Chem 41: 1439-1445.
3.Aoshima H, Kushida K, Takahashi M, et al, 1998 Circadian variation of urinary type I collagen crosslinked C-telopeptide and free and peptide-bound forms of pyridinium crosslinks. Bone 22: 73-78.
4.Gundberg CM, Markowitz ME, Mizruchi M, Rosen JF, 1985 Osteocalcin in human serum: a circadian rhythm. J Clin Endocrinol Metab 60: 736-739.
5.Greenspan SL, Dresner-Pollak R, Parker RA, Londo D, Ferguson L, 1997 Diurnal variation of bone mineral turnover in elderly men and women. Calcif Tissue Int 60: 419-423.
6.Gertz BJ, Clemens JD, Holland SD, Yuan W, Greenspan S, 1998 Application of a new serum assay for type I collagen cross-linked N-telopeptides: assessment of diurnal changes in bone turnover with and without alendronate treatment. Calcif Tissue Int 63: 102-106.
7.Hassanger C, Risteli J, Risteli L, Jensen SB, Christiansen C, 1992 Diurnal variation in serum markers of type I collagen synthesis and degradation in healthy premenopausal women. J Bone Miner Res 7: 1307-1311.
8.Heshmati HM, Riggs BL, Burritt MF, McAlister CA, Wollan PC, Khosla S, 1998 Effects of the circadian variation in serum cortisol on markers of bone turnover and calcium homeostasis in normal postmenopausal women. J Clin Endocrinol Metab 83: 751-756.
9.Ledger GA, Burritt MF, Kao PC, O’Fallon WM, Riggs BL, Khosla S, 1995 Role of parathyroid hormone in mediating nocturnal and age-related increases in bone resorption. J Clin Endocrinol Metab 80: 3304-3310.
10.Geoffriau M, Brun J, Chazot G, Claustrat B, 1998 The physiology and pharmacology of melatonin in humans. Horm Res 49: 136-141.
11.Ostrowska Z, Kos-Kudla B, Marek B, Swietochowska E, Gorski J, 2001 Assessment of the relationship between circadian variations of salivary melatonin levels and type I collagen metabolism in postmenopausal obese women. Neuro Endocrinol Lett. 22: 121-127.
12.Qvist P, Christgau S, Pedersen BJ, Schlemmer A, Christiansen C, 2002 Circadian variation in the serum concentration of C-terminal telopeptide of type I collagen (serum CTx): effects of gender, age, menopausal status, posture, daylight, serum cortisol, and fasting. Bone 31: 57-61.
13.Schlemmer A, Hassager C, 1999 Acute fasting diminishes the circadian rhythm of biochemical markers of bone resorption. Eur J Endocrinol 140: 332-337.
14.Henriksen DB, 2005 The gut feeling of bone remodeling IBMS BoneKEy 2: 16 -15.
15.Clowes JA, Robinson RT, Heller SR, Eastell R, Blumsohn A, 2002 Acute changes of bone turnover and PTH induced by insulin and glucose: Euglycemic and hypoglycemic hyperinsulinemic clamp studies. J Clin Endocrinol Metab 87: 3324-3329.
16.Chailurkit LO, Chanprasertyothin S, Rajatanavin R, Ongphiphadhanakul B, 2008 Reduced attenuation of bone resorption after oral glucose in type 2 diabetes. Clin Endocrinol (Oxf) 68: 858-862.
17.Yavropoulou MP, Tomos K, Tsekmekidou X, et al, 2011 Response of biochemical markers of bone turnover to oral glucose load in diseases that affect bone metabolism. Eur J Endocrinol 164: 1035-1041.
18.Henriksen DB, Alexandersen P, Bjarnason NH, et al, 2003 Role of gastrointestinal hormones in postprandial reduction of bone resorption. J Bone Miner Res 18: 2180-2189.
19.Clowes JA, Allen HC, Prentis DM, Eastell R, Blumsohn A, 2003 Octreotide abolishes the acute decrease in bone turnover in response to oral glucose. J Clin Endocrinol Metab 88: 4867-4873.
20.Schmidt WE, 1997 The intestine, an endocrine organ. Digestion 58: 56-58.
21.Vilsboll T, Krarup T, Sonne J, et al, 2003 Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab 88: 2706-2713.
22.Brown JC, Dryburgh JR, 1971 A gastric inhibitory polypeptide. II. The complete amino acid sequence. Can J Biochem 49: 867-872.
23.Ding KH, Zhong Q, Xu J, Isales CM, 2004 Glucose-dependent insulinotropic peptide: differential effects on hepatic artery vs. portal vein endothelial cells. Am J Physiol Endocrinol Metab 286: E773-779.
24.Fehmann HC, Goke R, Goke B, 1995 Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocrine Reviews 16: 390-410.
25.Knapper JM, Morgan LM, Fletcher JM, 1996 Nutrient-induced secretion and metabolic effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1. Proc Nutr Soc 55: 291-305.
26.Kogire M, Inoue K, Sumi S, Doi R, Takaori K, Yun M, 1988 Effects of synthetic human gastric inhibitory polypeptide on splachnic circulation in dogs. Gastroenterology 95: 1636-1640.
27.Yip RG, Boylan MO, Kieffer TJ, Wolfe MM, 1998 Functional GIP receptors are present on adipocytes. Endocrinology 139: 4004-4007.
28.Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI, 1993 Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 133: 2861-2870.
29.Kieffer TJ, McIntosh CH, Pederson RA, 1995 Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136: 3585-3596.
30.Xie D, Zhong Q, Ding KH, et al, 2007 Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone 40: 1352-1360.
31.Xie D, Cheng H, Hamrick M, et al, 2005 Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone 37: 759-769.
32.Bollag RJ, Zhong Q, Phillips P, et al, 2000 Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology 141: 1228-1235.
33.Pacheco-Pantoja EL, Ranganath LR, Gallagher JA, Wilson PJ, Fraser WD, 2011 Receptors and effects of gut hormones in three osteoblastic cell lines. BMC Physiol 11: 12.
34.Tsukiyama K, Yamada Y, Yamada C, et al 2006 Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Molecular Endocrinology 20: 1644-1651.
35.Ding KH, Shi XM, Zhong Q, et al, 2008 Impact of glucose-dependent insulinotropic peptide on age-induced bone loss. J Bone Miner Res 23: 536-543.
36.Zhong Q, Itokawa T, Sridhar S, et al, 2007 Effects of glucose-dependent insulinotropic peptide on osteoclast function. Am J Physiol Endocrinol Metab 292: E543-548.
37.McIntosh CH, Wheeler MB, Gelling RW, Brown JC, Pederson RA, 1996 GIP receptors and signal-transduction mechanisms. Acta Physiol Scand 157: 361-365.
38.Stanley KT, Van Dort C, Motyl C, Endres J, Fox DA, 2006 Immunocompetent properties of human osteoblasts: interactions with T lymphocytes. J Bone Miner Res 21: 29-36.
39.McClean PL, Irwin N, Hunter K, Gault VA, Flatt PR, 2008 (Pro(3))GIP[mPEG]: novel, long-acting, mPEGylated antagonist of gastric inhibitory polypeptide for obesity-diabetes (diabesity) therapy. Br J Pharmacol 155: 690-701.
40.Parker JC, Irwin N, Lavery KS, et al, 2007 Metabolic effects of sub-chronic ablation of the incretin receptors by daily administration of (Pro3)GIP and exendin(9-39)amide in obese diabetic (ob/ob) mice. Biol Chem 388: 221-226.
41.Hinke SA, Gelling RW, Pederson RA, et al, 2002 Dipeptidyl peptidase IV-resistant [D-Ala(2)] glucose-dependent insulinotropic polypeptide (GIP) improves glucose tolerance in normal and obese diabetic rats. Diabetes 51: 652-661.
42.Creutzfeldt W, 2005 The [pre-] history of the incretin concept. Regulatory Peptides 128: 87-91.
43.Baggio LL, Drucker DJ, 2007 Biology of incretins: GLP-1and GLP-2. Gastrenterology 132: 2131-2157.
44.Baggio LL, Drucker DJ, 2004 Clinical endocrinology and metabolism. GLP-1 and GLP-2. Best Pract Res Clin Endocrinol Metab 18: 531-534.
45.Yamada C, Yamada Y, Tsukiyama K, et al, 2008 The murine glucagonlike peptide-1 receptor is essential for control of bone resorption. Endocrinology 149: 574-579.
46.Lamari Y, Boissard C, Moukhtar MS, Jullienne A, Rosselin G, Garel JM, 1996 Expression of glucagon-like peptide 1 receptor in a murine C cell line: regulation of calcitonin gene by glucagon-like peptide 1. FEBS Lett 393: 248-252.
47.Crespel A, De Boisvilliers F, Gros L, Kervran A, 1996 Effects of glucagon and glucagon-like peptide-1-(7–36) amide on C cells from rat thyroid and medullary thyroid carcinoma CA-77 cell line. Endocrinology 137: 3674-3680.
48.Nuche-Berenguer B, Moreno P, Esbrit P, et al, 2009 Effect of GLP-1 treatment on bone turnover in normal, type 2 diabetic, and insulin-resistant states. Calcif Tissue Int 84: 453-461.
49.Nuche-Berenguer B, Moreno P, Portal-Nuñez S, et al, 2010 Exendin-4 exerts osteogenic actions in insulin-resistant and type 2 diabetic states. Regul Pept 159: 61-66.
50.Nuche-Berenguer B, Lozano D, Gutiérrez-Rojas I, et al, 2011 GLP-1 and exendin-4 can reverse hyperlipidic-related osteopenia. J Endocrinol 209: 203-210.
51.Sanz C, Vázquez P, Blázquez C, Barrio PA, Alvarez M del M, Blaїzquez E, 2010 Signaling and biological effects of glucagon-like peptide 1 on the differentiation of mesenchymal stem cells from human bone marrow. Am J Physiol Endocrinol Metab 298: E634–E643.
52.Nuche-Berenguer B, Portal-Nuñez S, Moreno P, et al, 2010 Presence of a functional receptor for GLP-1 in osteoblastic cells, independent of the cAMP-linked GLP-1 receptor. J Cell Physiol 225: 585-592.
53.Villanueva-Peñacarrillo ML, Delgado E, Trapote MA, et al, 1995 Glucagon-like peptide-1 binding to rat hepatic membranes. J Endocrinol 146: 183-189.
54.Bunck MC, Eliasson B, Corner A, et al, 2011 Exenatide treatment did not affect bone mineral density despite body weight reduction in patients with type 2 diabetes. Diabetes Obes Metab 13: 374-377.
55.Riedt CS, Cifuentes M, Stahl T, Chowdhury HA, Schlussel Y, Shapses SA, 2005 Overweight postmenopausal women lose bone with moderate weight reduction and 1 g/day calcium intake. J Bone Miner Res 20: 455-463.
56.Blonde L, Klein EJ, Han J, et al, 2006 Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes Obes Metab 8: 436-447.
57.Estall JL, Drucker DJ, 2006 Glucagon-like Peptide-2. Annu Rev Nutr. 26: 391-411.
58.Henriksen DB, Alexandersen P, Byrjalsen I, et al, 2004 Reduction of nocturnal rise in bone resorption by subcutaneous GLP-2. Bone 34: 140-147.
59.Henriksen DB, Alexandersen P, Hartmann B, et al, 2007 Disassociation of bone resorption and formation by GLP-2: a 14-day study in healthy postmenopausal women. Bone 40: 723-729.
60.Henriksen DB, Alexandersen P, Hartmann B, et al, 2009 Four-month treatment with GLP-2 significantly increases hip BMD: a randomized, placebo-controlled, dose-ranging study in postmenopausal women with low BMD. Bone 45: 833-842.
61.Askov-Hansen C, Jeppesen PB, Lund P, Hartmann B, Holst JJ, Henriksen DB, 2013 Effect of glucagon-like peptide-2 exposure on bone resorption: Effectiveness of high concentration versus prolonged exposure. Regul Pept 181: 4-8.
62.Nørholk LM, Holst JJ, Jeppesen PB, 2012 Treatment of adult short bowel syndrome patients with teduglutide. Expert Opin Pharmacother 13: 235-243.
63.Monami M, Dicembrini I, Antenore A, Mannucci E, 2011 Dipeptidyl peptidase-4 inhibitors and bone fractures: a meta-analysis of randomized clinical trials. Diabetes Care 34: 2474-2476.
Address for correspondence:
Maria P. Yavropoulou, MD, PhD, Endocrinologist, Division
of Endocrinology and Metabolism, Bone Metabolic Unit,
1 S. Kyriakidi Str., 54636, AHEPA University Hospital,
Thessaloniki, Greece, Tel.: +30 2310 993187,
Fax: +30 2310 994608, E-mail: margia@med.auth.gr
Received 28-07-2012, Accepted 11-01-2013