1Division of Endocrinology, Department of Medicine, 2Division of Oncology, Department of Medicine, 3Department of Pathology, 4Department of Neurosurgery, University of Mississippi Medical Center, 5GV (Sonny) Montgomery VA Medical Center, Woodrow Wilson, Jackson, Mississippi, USA
INTRODUCTION: Malignant prolactinoma is an exceedingly rare endocrine tumor and cannot be diagnosed on histological grounds alone. Similarly to other neuroendocrine tumors such as pheochromocytoma, the mitoses index, Ki-67, p53, and others are utilized in helping understand whether a tumor is benign or malignant or to better predict tumor behavior. We here present the unusual case of an unfortunate young man with an aggressive prolactinoma, the complications of which led to his premature death. CASE REPORT: A 25-year-old white man developed severe headaches, low energy, and decreased libido. A brain magnetic resonance imaging (MRI) showed a 4 x 3 x 2 cm pituitary tumor invading the left cavernous sinus. Laboratory findings revealed elevated prolactin (470 ng/mL) and adrenocorticotropic hormone (ACTH, 82 pg/ml) and decreased total testosterone (176 ng/dl). Visual fields showed superior quadrantanopia in the left eye. Transsphenoidal pituitary resection was undertaken. Pathology revealed a prolactinoma with atypical cells, diffuse p53 nuclear labeling, and a Ki-67 index of 23% (high). Postoperatively, prolactin remained elevated (725-891 ng/ml) and cabergoline was increased to 1 mg three times weekly, with serum prolactin further increasing to 3507 ng/ml five months postoperatively. Repeat MRI revealed extension of the tumor with optic chiasm compression and left orbit invasion. Because of acute left vision loss with ophthalmoplegia, an urgent left frontotemporal craniotomy and tumor resection were conducted. The Ki-67 index of the tumor was 24.8%, the mitotic figure immunostain phosphohistone-H3 positive. Sixty percent (60%) of tumor cells were positive for p53. Cabergoline was increased to 1 mg daily but prolactin remained elevated (770 ng/ml). The patient then underwent proton beam radiation to the area of concern involving the sella. Prolactin thereafter improved to 44 ng/ml. He then developed acute vision loss of the right eye with an MRI showing tumor in the right cavernous sinus. A 15 mm dural-based right temporal mass believed to be a metastasis was also noted. Following this scan, he was considered too high risk for debulking surgery and instead underwent gamma knife irradiation to the sella area. This shrank the right cavernous sinus tumor mass, while the right temporal mass increased in size. The patient developed blindness and left-sided weakness and required enteral feeding and tracheostomy after prolonged intubation. A trial of chemotherapy with temozolomide (350 mg daily for 5 days) near the end of his life was unsuccessful. He died on home hospice 31 months after his first surgery. CONCLUSION: Headaches, vision changes, and symptoms of androgen deficiency syndrome can be manifestations of an aggressive prolactinoma that might require surgery and additional medical therapy including cabergoline and temozolomide with an unpredictable time of survival.
Cabergoline, Pituitary carcinoma, Prolactin, Temozolomide
INTRODUCTION
Malignant
prolactinoma is exceedingly rare and cannot be diagnosed on histological
grounds alone. Similar to other neuroendocrine tumors such as pheochromocytoma,
the mitoses index, Ki-67, p53, and others are utilized in helping to determine
whether a tumor is benign or malignant or to better predict tumor behavior.
However, for pituitary tumors the presence of metastases within or outside the
CNS (distant from the pituitary) is required to characterize a tumor as
malignant or to ascertain that the tumor is not contiguous with the primary
sellar tumor,1,2 while the term "aggressive" should not be used
synonymously with "invasive".3 Pituitary carcinoma is very uncommon,
accounting for approx. 0.1% of all pituitary tumors. Treatment options are
limited. Survival is usually poor. We herein present the unusual case of an
unfortunate young man with an aggressive prolactinoma, the complications of
which led to his premature death.
CASE REPORT
A
25-year-old white man presented to his primary care physician with severe
headaches, low energy, and decreased libido, without clinical features of
Cushing's syndrome. A brain magnetic resonance imaging (MRI) showed a 4 x 3 x 2
cm pituitary tumor invading the left cavernous sinus (Figure 1). 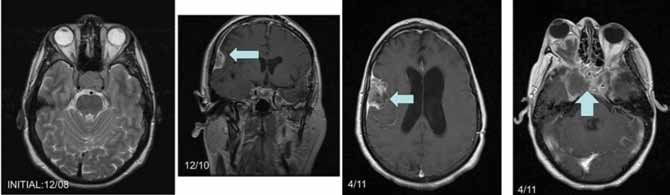
Figure 1. Serial magnetic resonance imaging of the pituitary / brain.
A. Initial MRI, horizontal cut, note sellar area
B. 2-year follow-up, vertical cut, note dural lesion right temporal
C. 28 months follow-up, emphasizing right temporal area
D. 28 months follow-up, emphasizing sellar area.
Laboratory
findings revealed elevated prolactin (diluted 470 ng/mL; normal less than 25
ng/ml), ACTH (82 pg/ml, normal, 10-60), serum cortisol of 19 mcg/dl, and
decreased total testosterone (176 ng/dl) with low normal LH and FSH. TSH, free
T4, and serum cortisol values were normal. Visual fields showed superior
quadrantanopia in the left eye. Transsphenoidal pituitary resection was
undertaken, taking into consideration the visual field defect and the potential
for plurihormonal tumor given the elevated ACTH and prolactin. Cabergoline or
bromocriptine were not administered preoperatively. Pathology revealed an
atypical pituitary adenoma with increased pleomorphism but no necrosis or
mitotic activity with stains diffusely positive for prolactin consistent with
prolactinoma (Figure 2). Note was made of atypical cells including
multinucleated giant cells and cells with bizarre, enlarged nuclei. The Ki-67
index was 23% (high) and there was diffuse p53 nuclear labeling.
Postoperatively, the patient was discharged on DDAVP (vasopressin), for
treatment of diabetes insipidus, and on hydrocortisone, as his morning serum
cortisol level was low at 3.6 mcg/dl with an ACTH of 6 pg/ml. Prolactin
remained elevated (725-891 ng/ml) and cabergoline was increased to 1 mg three
times weekly (Table 1). In spite of that, prolactin was 3507 ng/ml five months
postoperatively. Repeat MRI revealed extension of the tumor with optic chiasm
compression and left orbit invasion. Surgery was planned, but prior to this he
developed acute left vision loss with ophthalmoplegia and underwent urgent left
frontotemporal craniotomy and tumor resection at another institution. The Ki-67
labeling index of the tumor was 24.8%. The mitotic figure immunostain
phosphohistone-H3 was positive and 60% of tumor cells were positive for p53.
These outside pathology report parameters compared with the ones from the first
resection further document the progression of this pituitary tumor from
atypical adenoma to outright carcinoma. The patient's postoperative course was
complicated by expressive dysphagia and left CN III, IV, VI nerve palsies, and
ischemic brain pathology was attributed to possible intraoperative cerebral
vasospasm. Cabergoline was increased to 1 mg daily on discharge, and
levothyroxine and the antiepileptic drug Levetiracetam (Keppra) were added to
his regimen. Prolactin remained elevated (770 ng/ml). He then underwent proton
beam radiation (54 Gy in 30 fractions of 1.8 Gy per fraction) to the area of
concern involving the sella. Prolactin thereafter improved to 44 ng/ml. He
subsequently developed acute vision loss of the right eye (believed to be
related to ischemia/vasospasm) with an MRI showing tumor in the right cavernous
sinus (October 2010). A 15 mm dural-based temporal mass believed to be a
metastasis was also noted. Following this scan, he was considered too high risk
for debulking surgery and instead underwent gamma knife irradiation while on
dexamethasone therapy. This shrank the right cavernous sinus tumor mass, while
the right temporal mass increased in size (Figure 1, Dec 2010). The patient
developed blindness and left-sided weakness and required enteral feeding and
tracheostomy after a prolonged intubation. A trial of chemotherapy with
temozolomide (350 mg daily for 5 days as an inpatient)near the end of his life was unsuccessful. He died on home hospice
31 months after his first surgery. 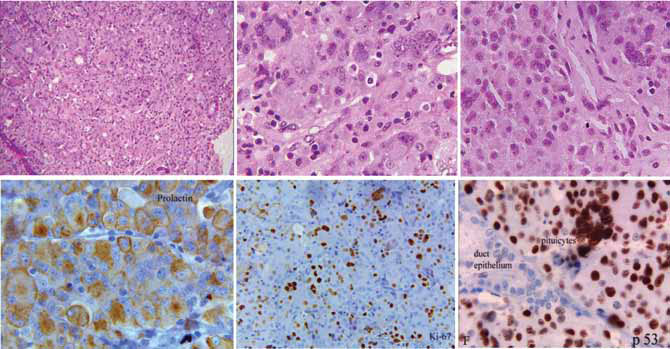
Figure 2. Histology (Hematoxylin & Eosin) and Immunohistochemistry (prolactin, Ki-67, p53) of atypical pituitary adenoma.
A. Acinar or organoid architecture replaced by sheets of cells (low power magnification)
B. Population of pleomorphic nuclei in this area of tumor, including multinucleated giant cells; the tumor cell nuclei have clumped
chromatin, huge nucleoli; there is no necrosis (high power magnification)
C. Sheets of cells with pleomorphic nuclei, mitoses are difficult to identify (medium power)
D. Tumor cells are diffusely positive for prolactin
E. High labeling index; 23% of nuclei are positive for Ki-67
F. Diffuse nuclear labeling with p53
DISCUSSION
Similarly
to other neuroendocrine tumors (NET) such as pheochromocytoma, the diagnosis of
pituitary carcinoma is made only after discovery of metastases or the presence
of tumor that is not contiguous with the primary sellar tumor.1-4
Proliferation markers Ki-67 labeling index (MIB-1), a monoclonal antibody
against the Ki-67 antigen, are used to predict tumor aggressiveness and
recurrence. A Ki-67 labeling index cutoff of 3% has been suggested to predict
invasiveness, with higher values being more invasive.3,5 Ki-67 is
more predictive in functioning tumors than in nonfunctioning tumor.4-6
Pituitary carcinomas average a higher Ki-67 index (11%), although there are
reports of carcinomas with a low Ki-67 index, suggesting that there are factors
other than proliferation that contribute to aggressiveness and malignant
potential. The presence of mitoses and nuclear pleomorphism in a pituitary
tumor, like the one presented here in our patient, should raise suspicion for
aggressive behavior, although studies differ as to whether these correlate with
the proliferation index.3,6 The World Health Organization
classification of endocrine tumors, including pituitary, requires specific
morphologic features and immunophenotypic characteristics (Ki-67 index, p53
staining), as those of our patient, in order to characterize a pituitary tumor
as atypical.5 Of note, the tumor in our patient already had p53
positivity and histologic features considered to be atypical at first
presentation and not only after radiation therapy. Therefore, it appears that
detailed histological subtyping represents an excellent predictor of aggressive
behavior in addition to analysis of such pituitary tumors for fibroblast growth
factor receptor 4, matrix metalloproteinases, pituitary tumor transforming
gene, Ki-67, p53, and deletions in chromosome 11.3
The
molecular pathogenesis of pituitary tumors is still largely unknown despite the
fact that several new genes have been identified in familial pituitary tumor
syndromes.7-11 Consideration of potential hereditary causes or
somatic GNAS mutation in pituitary tumorigenesis is important for initial
surgical planning and aftercare, including risk for recurrent disease.12
Recent studies have focused on identifying genetic differences between invasive
and noninvasive tumors. In a study using microarray analysis of nonfunctioning
pituitary adenomas, overexpression of 4 genes was confirmed in invasive tumors
(IGFBP5, MYO5A, FLT3, and NFE2L1) and, given the known effect of the product of
MYO5A on tumor cell migration and invasion, this may be an area of interest for
future research into tumor markers.7 Loss of heterozygosity through
allelic deletion at 11q13 (the site for the tumor-suppressor gene MEN1),
13q12-14, and at 10q and 1p has been implicated in some pituitary carcinomas.
However, as the number of pituitary tumors with described mutations or
deletions is small, it is difficult to infer this as a generalized mechanism
for aggressive tumor behavior.13 An important point to consider, as
in our patient, is the potential of radiation to induce further chromosomal
damage to an existing tumor but also normal cells, including pituicytes.
Prolactinomas make up approx. 50% of all pituitary tumors and respond to
dopamine agonist therapy, with reduction in size in up to 90% of (compliant)
patients.14 For prolactinomas that are resistant to conventional
doses of dopamine agonists, high dose cabergoline (12 mg per week or more),
surgery, and radiotherapy should be utilized or, if these fail, as in our
patient, additional therapies should be used including temozolomide.15,16
Pituitary carcinomas and NET can change their hormone expression profile, which
in part depends on their grade of differentiation.2,17 For instance,
von Hippel Lindau disease-associated pheochromocytomas are considered less
differentiated than pheochromocytomas in patients with multiple endocrine
neoplasia type 2 and have a more norepinephrine/normetanephrine prone
biochemical profile.18
Resistance
to cabergoline (dopamine D2-like receptor agonist) therapy, defined by the lack
of normalization of prolactin or failure of the tumor to shrink by 50%, occurs
in less than 10% of patients with prolactinoma.14 Histologically,
resistant prolactinomas have increased angiogenesis, cellular atypia,
proliferation, and invasiveness. There is a spectrum of resistance and most
resistant prolactinomas are not carcinomas. Mechanisms for resistance include
decreased D2 receptor gene transcription, decreased activity of receptors which
regulate D2 receptor expression, and decreased expression of the inhibitory G protein
which couples the D2 receptor to adenylyl cyclase.14,19 Given the
current healthcare state of affairs, one also has to take into account
noncompliance with medications that patients are unable to afford before
defining the respective tumor as medically refractory. Often, patients feel
ashamed to admit to not having taken their drugs because of financial concerns.
Treatment
for (truly) dopamine agonist resistant prolactinomas should include
transsphenoidal surgery and then radiation for persistent or recurrent disease,
although radiotherapy has limited success in these tumors. Advanced medical
therapy should be considered for those in whom these efforts fail.19
Temozolamide is an oral alkylating agent most often used to treat glioblastoma
multiforme which has been shown to be of benefit in some cases of resistant
pituitary tumors, including prolactinomas. (15,16,20,21) It interferes with DNA
replication via methylation of DNA guanine at the O-6 position. Low levels of
the DNA repair enzyme methylguanine methyltransferase (MGMT) may predict a more
favorable response to temozolomide (TMZ),22-24 although a recent
case series suggests otherwise and recommends against using this to guide
patient care. In this series and review of the literature, 5 of 6 prolactinomas
responded to TMZ, and 3 of 6 had low MGMT staining (2 of 6 with unknown MGMT
status).20
"Evidence"
for rare endocrine tumors is limited and composed of individual case studies.
This case highlights the morbidity and mortality associated with aggressive
pituitary tumors. Although survival is difficult to predict due to the small
number of reported cases, one review estimated survival at eight years from the
time of diagnosis and noted only 60% survival at one year after diagnosis of
metastasis.25
Further
elucidating the pathogenesis of aggressive pituitary tumors, including
prolactinomas, will help guide medical treatment options. Earlier diagnosis is
needed to commence aggressive treatment prior to the occurrence of metastases.
Although in our patient temozolamide was ineffective, it should be considered
in patients with aggressive pituitary tumors refractory to standard therapies.
REFERENCES
1. Heaney AP, 2011 Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab 96: 3649-3660.
2. Dudziak K, Honegger J, Bornemann A, et al, 2011 Pituitary carcinoma with malignant growth from first presentation and fulminant clinical course – case report and review of the literature. J Clin Endocrinol Metab 96: 2665-2669.
3. Mete O, Ezzat S, Asa SL, 2012 Biomarkers of aggressive pituitary adenomas. J Mol Endocrinol 49: R69-78.
4. Singer J, Koch CA, Kassahun W, et al, 2011 A patient with a large recurrent pheochromocytoma demonstrating the pitfalls of diagnosis. Nat Rev Endocrinol 7: 749-755.
5. DeLellis RA, Lloyd RV, Heitz PU, Eng C (eds), 2004 World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. Lyon: IARC Press.
6. Turner HE, Wass JA, 1999 Are markers of proliferation valuable in the histological assessment of pituitary tumours? Pituitary 1: 147-151.
7. Galland F, Lacroix L, Saulnier P, et al, 2010 Differential gene expression profiles of invasive and non-invasive non-functioning pituitary adenomas based on microarray analysis. Endocr Relat Cancer 17: 361-371.
8. Boikos SA, Stratakis CA, 2007 Molecular genetics of the cAMP-dependent protein kinase pathway and of sporadic pituitary tumorigenesis. Hum Mol Genet 16: R80-87.
9. Stratakis CA, Tichomirowa MA, Boikos S, et al, 2010 The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet 78: 457-463.
10. Ahmad S, Aaltonen LA, Georgitsi M, et al, 2007 Do single nucleotide polymorphisms in the AIP gene and MEN 1 gene predispose individuals to the development of familial isolated pituitary tumors ? Exp Clin Endocrinol 115: S35-36.
11. Glasker S, Vortmeyer AO, Lafferty AR, et al, 2011 Hereditary pituitary hyperplasia with infantile gigantism. J Clin Endocrinol Metab 96: E2078-2087.
12. Vortmeyer AO, Glasker S, Mehta GU, et al, 2012 Somatic GNAS mutation causes widespread and diffuse pituitary disease in acromegalic patients with McCune-Albright syndrome. J Clin Endocrinol Metab 97: 2404-2413.
13. Gurlek A, Karavitaki N, Ansorge O, Wass JA, 2007 What are the markers of aggressiveness in prolactinomas? Changes in cell biology, extracellular matrix components, angiogenesis, and genetics. Eur J Endocrinol 156: 143-153.
14. Molitch ME, 2008 The cabergoline-resistant prolactinoma patient: new challenges. J Clin Endocrinol Metab 93: 4643-4645.
15. Byrne S, Karapetis C, Vrodos N, 2009 Novel use of temozolomide in a patient with malignant prolactinoma. J Clin Neurosci 16: 1694-1696.
16. Neff L, Weil M, Hedges TR, et al, 2007 Temozolomide in the treatment of an invasive prolactinoma resistant to dopamine agonists. Pituitary 10: 81-86.
17. Miehle K, Tannapfel A, Lamesch P, et al, 2004 Pancreatic neuroendocrine tumor with ectopic adrenocorticotropin production upon second recurrence. J Clin Endocrinol Metab 89: 3731-3736.
18. Koch CA, Mauro D, Walther MM, et al, 2002 Pheochromocytoma in von hippel-lindau disease: distinct histopathologic phenotype compared to pheochromocytoma in multiple endocrine neoplasia type 2. Endocr Pathol 13: 17-27.
19. Oh MC, Aghi MK, 2011 Dopamine agonist-resistant prolactinomas. J Neurosurg 114: 1369-1379.
20. Dillard TH, Gultekin SH, Delashaw JB Jr, Yedinak CG, Neuwelt EA, Fleseriu M, 2011 Temozolomide for corticotroph pituitary adenomas refractory to standard therapy. Pituitary 14: 80-91.
21. Kovacs K, Horvath E, Syro LV, et al, 2007 Temozolomide therapy in a man with an aggressive prolactin-secreting pituitary neoplasm: Morphological findings. Hum Pathol 38:185-189.
22. Bush ZM, Longtine JA, Cunningham T, et al, 2010 Temozolomide treatment for aggressive pituitary tumors: correlation of clinical outcome with O(6)-methylguanine methyltransferase (MGMT) promoter methylation and expression. J Clin Endocrinol Metab 95: E280-290.
23. Raverot G, Sturm N, de Fraipont F, et al, 2010 Temozolomide treatment in aggressive pituitary tumors and pituitary carcinomas: a French multicenter experience. J Clin Endocrinol Metab 95: 4592-4599.
24. Syro LV, Ortiz LD, Scheithauer BW, et al, 2011 Treatment of pituitary neoplasms with temozolomide: a review. Cancer 117: 454-462.
25. Kars M, Roelfsema F, Romijn JA, Pereira AM, 2006 Malignant prolactinoma: case report and review of the literature. Eur J Endocrinol 155: 523-534.
Address for correspondence:
Prof. Dr. med. habil. Christian A. Koch, FACP, FACE, Head, Endocrine Tumor Program, Cancer Institute, Director, Division of Endocrinology, University of Mississippi Medical Center, 2500 North State Street, University of Mississippi Medical Center, Jackson, MS 39216, USA, Tel.: +1-601-984-5495,
Fax: +1-601-984-5769, e-mail: ckoch@umc.edu
Received 01-06-12, Revised 19-08-12, Accepted 11-09-12