1Department of Pediatric Endocrinology, “P. & A. Kyriakou” Children’s Hospital, Athens, Greece, 2Pediatric Endocrine Unit, Department of Pediatrics, Nicosia, Cyprus
There have been considerable advances concerning understanding of the early and later stages of ovarian development; a number of genes have been implicated and their mutations have been associated with developmental abnormalities. The most important genes controlling the initial phase of gonadal development, identical in females and males, are Wilms’ tumor suppressor 1 (WT1) and steroidogenic factor 1 (SF1). Four genes are likely to be involved in the subsequent stages of ovarian development (WNT4, DAX1, FOXL2 and RSPO1), but none is yet proven to be the ovarian determining factor. Changes in nomenclature and classification were recently proposed in order to incorporate genetic advances and substitute gender-based diagnostic labels in terminology. The term “disorders of sex development” (DSD) is proposed to substitute the previous term “intersex disorders”. Three main categories have been used to describe DSD in the 46,XX individual: 1) disorders of gonadal (ovarian) development: ovotesticular DSD, previously named true hermaphroditism, testicular DSD, previously named XX males, and gonadal dysgenesis; 2) disorders related to androgen excess (congenital adrenal hyperplasia, aromatase deficiency and P450 oxidoreductase deficiency); and 3) other rare disorders. In this mini-review, recent advances concerning development of the genital system in 46,XX individuals and related abnormalities are discussed. Basic embryology of the ovary and molecular pathways determining ovarian development are reviewed, focusing on mutations disrupting normal ovarian development. Disorders of sex development according to the revised nomenclature and classification in 46,XX individuals are summarized, including genetic progress in the field.
Disorders of sex development, Embryology of the ovary, Ovarian development, Congenital adrenal hyperplasia, Androgen excess, Gonadal dysgenesis
INTRODUCTION
There is a growing body of knowledge related to the genes that control sex determination and differentiation. In the initial stage of gonadal development the genetic control does not differ in either gender. Regulatory genes controlling the development of the genital ridge and the formation of the bipotent gonad have been identified and developmental anomalies resulting from gene mutations have been described.1 The differentiation of the bipotent gonad to an ovary or testis follows and is also under genetic control. Several genes affecting testicular differentiation have been determined, whereas very little is known about ovarian formation. Since a functioning ovary is not necessary for female phenotype development, while a testis is necessary for the male phenotype, the development of the ovary has been incorrectly considered a development by ‘default’. Since 2004, new findings have suggested that specific genes are required for the early development of the ovary and that mutations in these genes influence ovarian development and result in specific clinical syndromes.2
In 2006, the European Society for Pediatric Endocrinology (ESPE) and the Lawson Wilkins Pediatric Endocrine Society (LWPES) reviewed the overall management of intersex disorders and proposed changes in terminology3 (Table 1 ). Given the significant advances in the understanding of molecular causes of abnormal sexual development, it became necessary to integrate current knowledge with the classification of intersex disorders. Furthermore, there has been dissatisfaction about existing nomenclature of intersex disorders among both health professionals and patients as to the gender-based diagnostic labels.3 The term ‘disorders of sex development’ (DSD) is now proposed to define congenital conditions in which a dysharmony between chromosomal, gonadal and anatomical sex exists.3 A new classification system for the causes of DSD has been proposed based on the karyotype. This terminology, however, also includes the term “sex” in the description of the specific developmental abnormality with the inevitable associated connotation.4
The present mini-review focuses on the 46,XX individual, with emphasis on recent advances in knowledge pertaining to ovarian development and related abnormalities. In the first part, basic embryology of the ovary, genetic control of ovarian development and relevant mutations are reviewed. In the second part, the DSD in the 46,XX individual according to the revised nomenclature and classification are discussed. The latter part aims to familiarize the clinician with recent changes in terminology and to suggest how the new terms may be incorporated in everyday practice.
BASIC EMBRYOLOGY OF THE OVARY AND DUCTS
The urogenital ridge, from which the urogenital system will derive, arises at approximately the 4th week of gestation in the intermediate mesoderm.5 The indifferent gonad, identical in females and males, emerges on the ventromedial surface of the mesonephros as a derivative of the intermediate mesoderm. The indifferent or bipotent gonad is formed by proliferation of the coelomic epithelium and a condensation of mesenchymal cells of mesonephric origin.6 Primordial germ cells (PGCs) derive from the epiblast, the outer ectodermal layer of the embryo; they subsequently move to the yolk sac wall and then migrate along the dorsal mesentery of the hind gut to the gonadal ridge (Figure 1).1 During migration, PGCs undergo cell division and, once in the genital ridge (by the end of the 5th week), lose their motility, begin to aggregate and continue to proliferate by mitosis.
Figure 1. The gonad emerges on the ventromedial surface of the mesonephros at the 4th week of gestation. The migration of the primordial germ cells from the wall of the yolk sac along the dorsal mesentery of the hind gut to the gonadal ridge is shown as blue circles (reproduced and modified by permission of Professor M. Kouloukoussa, Dept of Embryology and Histology, University of Athens).
In the female embryos, PGCs differentiate to oogonia and continue to divide by mitosis. Shortly before and during the arrival of PGCs, the epithelium of the genital ridge proliferates and the epithelial cells penetrate the underlying mesenchyme forming the primitive sex cords,6 which surround the oogonia. During the 7th week the proliferating epithelium gives rise to a second generation of cords, the cortical cords.6 At the 10th week some oogonia will arrest their division and differentiate to oocytes. In the 4th month the cortical cords split into clusters surrounding one or more of the oocytes and the earliest primary follicles appear (Figure 2). The oocytes increase rapidly in number and by the 5th month of gestation the total number of oocytes in the ovary reaches its maximum. However, most oocytes undergo apoptosis, their number diminishes and many follicles become atretic. At birth, the total number of oocytes is estimated to range from 600,000 to 800,000; subsequently the majority of follicles will become atretic and at the beginning of puberty approximately 400,000 follicles will remain and less than 500 will proceed to ovulation.7 
Figure 2. Ovary and genital ducts at the 5th month. The medullary cords are degenerating, the cortical zone of the ovary contains groups of oogonia surrounded by follicular cells (reproduced by permission).6
In the bipotent embryonic stage both M?llerian (paramesonephric) and Wolffian (mesonephric) ducts are present. Müllerian ducts are formed by an invagination of a tube from the surface coelomic epithelium of the mesonephros.8 In females, the Wolffian duct regresses and the Müllerian duct differentiates into oviduct, uterus and upper vagina.
MOLECULAR PATHWAYS OF SEX DETERMINATION IN THE 46,XX INDIVIDUAL
Genes important for the formation of the bipotent gonad
Genes important for the initial formation of the genital ridge include Wilm’s tumor suppressor 1 (WT1) and steroidogenic factor 1 (SF1).1
The WT1 gene located on chromosome 11p13 (OMIM#67102) is expressed in the urogenital ridge, mesonephros, kidney, gonad and in the granulosa cells in females. It encodes a transcriptional factor playing a role in gonadal development.1,8 Mutations in the WT1 gene occurring in the XY genotype are associated with both gonadal dysgenesis and renal anomalies (Frasier Syndrome, Deny’s-Drash syndrome and WAGR syndrome).1,9 By contrast, the same mutations of the WT1 gene in the XX individual have no effect on gonadal development, thus resulting in a normal female with associated renal anomalies (focal segmental sclerosis) and a predisposition to Wilm’s tumor.10,11
Another important gene in early gonadal development is SF1 (located on 9q33, OMIM #184757), which is expressed in the developing urogenital ridge, hypothalamus, the anterior pituitary gland and the adrenal glands.12 It encodes a transcription factor regulating the expression of a number of genes that participate in sexual development, including all the cytochrome P-450 steroid hydroxylase enzymes and the 3β-hydroxysteroid dehydrogenase. SF1 knockout mice fail to develop adrenal glands and gonads and die at birth.13 In humans, heterozygous mutations in SF1 can lead to adrenal and gonadal failure in XY individuals with a variety of phenotypic combinations, including adrenal failure with variable degrees of testicular failure and normal adrenal function with complete sex reversal.14,15 Heterozygous partial loss of function mutations in SF1 have been described in a cohort of boys with bilateral anorchia (vanishing testis syndrome) and in 46,XY patients with severe undervirilization without adrenal insufficiency.16,17 Thus the phenotypic spectrum of genetic defects in the SF1 gene is broad and depends on the specific mutation.18 It ranges from complete testicular dysgenesis with maintenance of Müllerian structures to severe penoscrotal hypospadias with or without adrenal insufficiency. A heterozygous mutation of SF1 was detected in a 46,XX prepubertal individual presenting with adrenal insufficiency and apparently normal ovarian differentiation.19
A number of other genes are required to achieve normal development of the bipotent gonad in mice. These include empty-spiracles homeobox gene 2 (EMX2), a member of the polycomb group M33, and Lim homeobox gene 9 (LHX9). Mutations in the genes encoding these proteins have not yet been associated with gonadal abnormalities in humans.8
Genes involved in the development of the ovary
Despite extensive investigation with regard to ovarian development, the number of related genes so far identified is still limited. Four genes (WNT4, DAX1, FOXL2 and RSPO1) are likely to be involved in the early development of the ovary (Figure 3). However, despite their importance in the development of the ovary, none has been proven to be the ovarian determining factor. 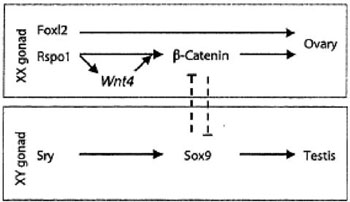
Figure 3. Genes likely involved in the development of the ovary and testis.
WNT4 (wingless-related mouse mammary tumor virus integrated site 4), located on chromosome 1p36.23-p35.1 (OMIM#603490), is expressed in the mesonephric mesenchyme, the coelomic epithelium, the female gonad and the mesenchyme surrounding the Müllerian ducts.8 In mice, WNT4 is important for the development of several organs, including the kidney, fallopian tubes, uterus and ovaries, as well as the mammary glands. WNT4 suppresses testosterone biosynthesis in the female mouse, inhibits the formation of the male-specific vascularization in the ovary and represses migration of endothelial and steroidogenic cells from the mesonephros and adrenal anlagen to the ovary, this suggesting an anti-testis and pro-ovary function.20-22 Furthermore, WNT4 is likely to play a role in follicle development and maturation.23,24 WNT4 seems to exert its functions via the upregulation of follistatin, a signaling molecule that binds to members of the transforming growth factor β family of proteins, as suggested by expression and null mutation analysis.21 In humans, female patients carrying a heterozygous mutation of the recently identified WNT4 gene present M?llerian duct abnormalities along with clinical and biological evidence of hyperandrogenism, implying that WNT4 gene plays an important role in the development of the female phenotype.24-26
DAX1 (dosage sensitive sex reversal adrenal hypoplasia congenital critical region of the X chromosome gene1), a gene located on chromosome Xp21.3-p21.2 (ΟMIM#300473), encodes a peculiar nuclear receptor, lacking a classic DNA-binding domain, is considered to play a role in both testicular and ovarian development.27,28 DAX1 mutations are associated with X-linked primary adrenal insufficiency and hypogonadotropic hypogonadism.29 Duplication of the region coding for this gene caused 46,XY individuals to develop as females, a situation that led to the hypothesis that DAX1 might be the ovary determining gene.28 However, its inactivation in mice does not impair ovarian development or other aspects of female differentiation, while it impairs spermatogenesis, suggesting that DAX1 is not an ovarian determining gene but rather plays a critical role in spermatogenesis.30 DAX1 acts as an anti-SRY factor in the process of gonadal sex differentiation and is upregulated by WNT4, its activation being mediated via the WNT/β-catenin pathway.31
FOXL2 is a member of a large family of folkhead/winged helix transcriptional factors. The FOXL2 gene is located on chromosome 3q23 (OMIM#605597). It is expressed in the gonads, pregranulosa and later in granulosa cells and is essential for granulosa cell differentiation and ovary maintenance.32 FOXL2 plays a role in the XX sex reversal phenotype of the polled intersex syndrome in goats, characterized by complete female to male sex reversal.33 More than 130 mutations in FoxL2 have been associated with a human congenital disease, the blepharophimosis-ptosis-epicanthus inversus syndrome (BPES).34 Mutations leading to a significantly shortened FOXL2 protein often cause BPES type I, which is characterised by eyelid abnormalities and premature ovarian failure, thus indicating a functional role in ovarian development or maintenance.35 Interestingly, inducible deletion of FOXL2 in adult ovarian follicles in the mouse model leads to immediate upregulation of testis-specific genes, including the critical SRY target gene SOX9 (located on chromosome 17q24.3-q25.1, OMIM#608160).36 It has been shown that ablation of FOXL2 results in somatic sex reprogramming of adult ovaries leading to testis development, thus implying that FOXL2 has an additional crucial role in maintaining femaleness, at least in mice.
R-spondins are a recently characterized small family of growth factors which are thought to play an essential role in ovarian development. R-spondins interact with β-catenin and may also synergize with WNT proteins,37 possibly through positive regulation of WNT4 signaling.38 R-spondin1 and FOXL2 act in two distinct cellular types during goat ovarian differentiation.39 This interaction appears critical for early genital development and ovarian determination. Mutations in the R-spondin1 (RSPO1) gene have been associated with 46,XX testicular DSD in the absence of the testis-determining gene SRY, as well as with ovotesticular DSD in a 46,XX individual.40,41 Palmoplantar hyperkeratosis, predisposition to squamous cell carcinoma of the skin, congenital bilateral corneal opacities, onychodystrophy and hearing impairment were additional findings in such cases.40,41 Therefore, the RSPO1 gene (located on chromosome 1p34.3, OMIM#609595) appears to be directly involved in ovarian determination.
DISORDERS OF SEX DEVELOPMENT (DSD) IN THE 46,XX INDIVIDUAL (TABLE 2 )
A brief description of each condition is described below, with special emphasis on the pathophysiology and relevant advances in genetics.
A. Disorders of gonadal (ovarian) development
1. Ovotesticular DSD, previously named true hermaphroditism. It is a very rare disorder defined by the presence of both ovarian and testicular tissue in the same individual (Figure 4). In infancy the gonads appear to have normal ovarian tissue with numerous follicles and normal testicular tissue with seminiferous tubules containing germ cells. However, as time passes the ovarian tissue usually becomes functional and the testicular tissue regresses, becoming dysgenetic and showing interstitial fibrosis and lack of spermogonia.42,43 Besides the presence of ovotestis, ovary and testis may be found separately.42 Ovarian tissue is most often found on the left side and testicular tissue on the right side.42 There may be a mixture of Müllerian and Wolffian derivatives; in most patients uterus or unicornus uterus is observed. The external genitalia are ambiguous with various degrees of virilization, while, rarely, normal female genitalia may be present.44,45 The SRY gene (located on chromosome Yp11.3, OMIM#480000) in 46,XX ovotesticular patients is present in approximately 1/3 of the cases.43,44 In the SRY negative cases the mechanism responsible for the presence of testicular tissue is unclear. It may be that some patients have Y sequences confined to testicular tissue (therefore escaping detection via analysis of leucocyte DNA), there may be autosomal or X linked mutations in unknown genes controlling testicular development or there may have been an early presence of SRY sufficient to differentiate testicular tissue but vanishing later on.43,46 A mosaicism with a Y bearing cell line in the gonad is another possibility, as the SRY gene is expressed in the ovotestes of XX ovotesticular DSD.47 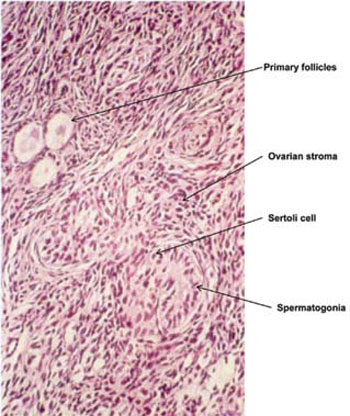
Figure 4. Histology of the gonad of a 46,XX ovotesticular DSD neonate showing a part of the gonad containing primary follicles and another part containing testicular tissue with spermatogonia (from the Pediatric Endocrinology Department of the “P. & A. Kyriakou” Children’s Hospital in Athens).
2. Testicular DSD (SRY+, dup SOX9, RSPO1), previously named XX males or XX sex reversal
It is a rare syndrome affecting 1 in 20,000-25,000 newborn males.48 The majority of the cases are phenotypically normal males but in some cases there is genital ambiguity. Approximately 10-15% show various degrees of hypospadias.49 Subjects with testicular 46,XX DSD differ from 46,XX ovotesticular individuals, as they have no ovarian tissue. They are often short, with azoospermia and with a high incidence of maldescended testes.50 They often display hypogonadism and gynaecomastia. Y chromosomal material including the SRY gene is detected in 90% of the cases. Y sequences are usually translocated to the distal tip of the short arm of the paternal X chromosome or to an autosome.48,49,51 However, in 10% of subjects with testicular DSD, SRY is negative, although patients present with different degrees of masculinization. The etiology of sex reversal in these cases is still unclear: mutations in a yet unknown X-linked or autosomal gene involved in testis differentiation or a hidden Y chromosome mosaicism limited to the gonad have been suggested.52 A newborn infant with severe penile/scrotal hypospadias and 46,XX,dup(17)(q23.1q24.3)/46,XX karyotype has been described.53 The duplicated SOX9 gene was on the maternally derived rearranged chromosome 17, suggesting that the SOX9 gene is important for testis formation and its duplication is sufficient to initiate testis differentiation in the absence of SRY. 53 Partial duplication of chromosome 22q (duplication of bands from 22q11.2-22q13) or overexpression of the SOX10 gene at 22q13 (OMIM#602229) can also lead to testicular DSD in 46,XX subjects in the absence of SRY.54 Furthermore, mutations in the RSPO1 gene, as previously mentioned, have been associated with testicular DSD in 46,XX subjects, in the absence of SRY, together with palmoplantar hyperkeratosis and predisposition to squamous cell carcinoma of the skin.40 However, with the exception of these few cases, SRY negative testicular DSD remains largely unexplained. The presence of familial 46,XX testicular DSD and familial 46,XX ovotesticular DSD in the same pedigree remains unexplained.55 These familial cases, in which XX ovotesticular DSD coexist with testicular DSD in the same sibship, provide evidence to support the hypothesis that these disorders constitute variable manifestations of the same genetic defect with differences in the expression and penetrance of the mutant gene.56 It should also be mentioned that an unknown interplay between several genes could be modified by still unidentified environmental factors, known as endocrine disruptors.57
3. Gonadal dysgenesis
In gonadal (ovarian) dysgenesis with normal XX karyotype, patients present with a female phenotype but fail to proceed to puberty and do not develop female secondary characteristics. They have elevated gonadotrophins and streak gonads.58 The streak gonads are similar to those found in patients with Turner syndrome; however, patients with gonadal dysgenesis do not have short stature or other stigmata associated with Turner syndrome.58,59 The condition is heterogeneous, most likely comprising several different etiological entities. In many cases it is inherited as an autosomal recessive trait, while the rest of the cases appear sporadic.59 In some forms the defect is restricted to the gonads, whereas in others affected females show neurosensory hearing loss (Perrault syndrome).60,61 In a subset of patients with ovarian dysgenesis, mutations in the FSH receptor gene were detected.62 Gonadal dysgenesis has also been associated with major X chromosome abnormalities. Genetic studies in such cases have identified several loci at Xq (including the BMP15 gene, the FMR1 gene and possibly the QM gene) and Xp11.2-p.22.1 whose function is related to ovarian development.63-66 Other chromosomal abnormalities such as 47,XXX karyotype and Xq deletions more often cause pubertal arrest, as a result of premature ovarian failure, than absent pubertal development.67,68
B. Androgen excess, previously named female pseudohermaphroditism
1. Fetal (congenital adrenal hyperplasia, glucocorticoid receptor mutations)
Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH), a group of autosomal recessive disorders of cortisol biosynthesis with variable genetic background, is one of the most common inherited metabolic disorders, the clinical consequences of which include genital ambiguity.69
Three forms of CAH lead to ambiguity of the external genitalia in 46,XX patients: 21-hydroxylase deficiency, 11-hydroxylase deficiency and 3β-hydroxysteroid dehydrogenase 2 deficiency. Mutations in enzymes involved in adrenal steroid biosynthesis lead to glucocorticoid deficiency, with consequent increase in ACTH, resulting in adrenal androgen excess and adrenal hyperplasia. Female patients with CAH have intact female internal genitalia. The formation of the external genitalia from the common primordia is completed by the 16th week. Thus, increased androgens secreted from the adrenal glands in the female embryo with CAH during this critical period causes virilization of the external genitalia (Figure 5). The excess of the adrenal androgen leading to virilization of the external genitalia in the newborn females also leads to accelerated growth and hyperandrogenemia later in life in patients not adequately treated. 
Figure 5. a. The 46,XX neonate with congenital adrenal hyperplasia should be distinguished from b. the XY neonate with partial androgen insensitivity syndrome and palpable testes. c. The 46,XX neonate with congenital adrenal hyperplasia with complete fusion of the labioscrotal folds and penile urethra should be distinguished from d. the XY neonate with bilateral cryptorchidism and hypospadias. (From the Pediatric Endocrine Unit in Nicosia, Cyprus).
21-hydroxylase deficiency (CYP21A2) accounts for approximately 90-95% of all cases of CAH. The classic form of 21-hydroxylase deficiency occurs in about 1 in 5,000 to 1 in 15,000 live births, whereas heterozygous mutations of the 21-hydroxylase gene occur in approximately 1 in 60.70,71 The CYP21A2 enzyme is a P450 type II enzyme located on the microsomes and requiring electron transfer from NADPH via the electron donor enzyme P450 oxidoreductase (POR). CYP21A2 is encoded by the 21-hydroxylase gene (CYP21A2) located on chromosome 6 (6p21.3) (OMIM#201910).
21-hydroxylase deficiency leads to an accumulation of 17-hydroxyprogesterone (17OHP) and consequently to an increase of 21-deoxycortisol and Δ4 androstenedione (Δ4 Α). Assessment of 17OHP, Δ4 A and 21-deoxycortisol can be useful markers for diagnosing and for follow-up of the patients,72 although the latter is not commonly used. The major adrenal androgens, androstendione and DHEA can be metabolized into testosterone and DHT (dihydrotestosterone), resulting in masculinization of the female fetus.58
The classic form of 21-hydroxylase deficiency presents either as the simple virilizing (non-salt losing) or the salt-losing form, reflecting the degree of cortisol and aldosterone deficiency.71 Female infants with classic 21-hydroxylase deficiency have ambiguous genitalia of various degree. The salt-losing form of 21-hydroxylase deficiency also presents with symptoms related to glucocorticoid and aldosterone deficiency. A life-threatening adrenal crisis (‘salt-losing crisis’), due to severe renal salt loss, may occur in the neonatal period usually between the 7th and 21st days of life.70 The milder, nonclassic or late-onset form of 21-hydroxylase deficiency is more frequent (from 1:100 to 1:500 in various Caucasian populations) than the classic form. Most girls are asymptomatic at birth and may present later on with premature pubarche during childhood, hirsutism and/or menstrual irregularities during adolescence, subfertility in later life or a phenotype resembling polycystic ovary syndrome.71,73,74 Several mutations in the CYP21A2 gene have been described with a broad overlap with regard to the degree of virilization and a variable genotype-phenotype correlation.The concordance between phenotype and genotype varies in the different forms, being 100% in the salt-losing variety.70,75
11β-hydroxylase deficiency (CYP11Β1) represents about 5-8% of CAH cases and occurs in 1 out of 200,000 live births.70 It is caused by mutations in the 11β-hydroxylase gene, located on chromosome 8q21 (ΟMIM #610613). 11β-hydroxylase catalyses the conversion of 11-deoxycortisol (S) to cortisol and the conversion of 11-deoxycorticosterone (DOC) to corticosterone.70 11β-hydroxylase deficiency leads to decreased cortisol secretion, accumulation of 11-deoxycortisol and DOC, resulting in significant hypertension. Additionally, increased production of adrenal androgens results in virilization of the external genitalia in newborn females.58 A nonclassic form of 11OHD has also been described in female patients born with normal genitalia and signs of androgen excess during childhood or adulthood.76
3β-hydroxysteroid dehydrogenase (HSD3B2) type 2 deficiency is the least common form, representing less than 2% of all CAH cases.58 There are two isoforms of 3β-hydroxysteroid dehydrogenase, 3βHSD type 1 and 3βHSD type 2, which are encoded by the HSD3B1 and HSD3B2 genes, respectively.70 The HSD3B2 gene is located on chromosome 1 (1p13.1, OMIM #201810) and is mainly expressed in the adrenal and the gonad.70 3βHSD catalyses steroid synthesis via three pathways, the conversion of the Δ5 steroids (Pregnenolone, 17-hydroxypregnenolone (17Preg) and DHEA) to the Δ4 steroids (Progesterone, 17OHP and Androstenedione, respectively), thereby affecting all three biosynthetic pathways (mineralcorticoids, glucocorticoids, sex steroids). Δ5-steroids (DHEA, 17Preg and their metabolites) are raised and there is a high ratio of Δ5 to Δ4 steroids.
Classic 3 beta-HSD deficiency is a rare form of congenital CAH that impairs steroidogenesis in both the adrenals and gonads causing varying degrees of salt loss in both sexes and incomplete masculinization of the external genitalia in genetic males. Female patients are not severely masculinized during fetal life due to the weak androgenic actions of DHEA and may present with clitoridomegaly or even normal external genitalia. The clinical spectrum is broad and includes a variable disease expression; a severe salt-wasting, simple virilization or a nonclassic phenotype (hirsutism and menstrual irregularities).70 In the non-classic form of the disease, women often present with infertility because of oligo-ovulation, which is reversed by dexamethasone.
P450 oxidoreductase deficiency (POR) is a novel form of CAH, classified as a DSD of fetoplacental androgen excess. It is not caused by mutations in genes encoding steroidogenic enzymes but in a gene encoding for an electron donor enzyme, therefore indirectly affecting several enzymes involved in steroidogenesis, including CYP17A1, CYP21A2 and CYP19A1 which require electron transfer from NADPH via POR for catalytic activity.70,77 The POR gene is located on the long arm of chromosome 7 (7q11.2, OMIM#124015). POR deficiency leads to partial deficiency of both 17α-hydroxylase and 21-hydroxylase activities. 17OHP is raised, but not to the extent observed in 21-hydroxylase deficiency. DHEA, DHEAS and androstendione are low to normal because of the partial 17α-hydroxylase deficiency. In most cases there is no mineralcorticoid deficiency.77 Basal values of cortisol are usually normal or nearly normal but do not respond normally to ACTH stimulation, indicating chronically compensated adrenal insufficiency. ACTH values may be high.
Affected girls may present with significant virilization of the external genitalia, indicating prenatal androgen excess.70,77 However, there is no progression of postnatal virilization and circulating androgen concentrations are low or low normal. Maternal virilization may occur, manifesting with acne, hirsutism and sometimes voice deepening usually around mid-gestation, followed by prompt reversal after delivery. In order to explain the paradox of prenatal androgen excess (virilized genitalia in affected girls and maternal virilization during pregnancy) and the finding of low or low normal circulating androgens in affected patients postnatally, an alternative pathway towards androgen synthesis, present only during fetal life, has been proposed.78
During early adolescence, patients may present with primary amenorrhea, polycystic ovaries or large ovarian cysts with a tendency to rupture. Affected patients may also have skeletal malformations, including craniofacial malformations, craniosynostosis, arachnodactyly, clinodactyly, radiohumeral synostosis and bowed femora, the pathogenesis of which remains unknown.70,77 Given that most drugs are metabolized by a number of hepatic P450 enzymes, there is a potential for altered drug metabolism of important clinical consideration in these patients, which is under assessment.77
Glucocorticoid receptor mutations
Glucocorticoid resistance is a rare familial or sporadic disorder characterized by increased cortisol secretion without clinical evidence of hypercortisolism but with manifestations of androgen and mineralcorticoid excess.79,80 Mutations of the glucocorticoid receptor gene cause inadequate transduction of the glucocorticoid signal in glucocorticoid target tissues and compensatory elevations in circulating cortisol and ACTH concentrations. Although adequate compensation is achieved by the elevated cortisol concentrations, in the majority of patients with this disorder, the excess ACTH secretion often results in increased production of adrenal steroids with androgenic and/or mineralcorticoid activities. The clinical spectrum of the condition is broad, ranging from completely asymptomatic to severe hyperandrogenism, fatigue and/or hypertension with or without electrolyte abnormalities. Glucocorticoid resistance may also be the cause of ambiguous genitalia in female infants who have elevated cortisol levels at baseline and after dexamethasone.79
2. Fetoplacental (aromatase deficiency)
Aromatase deficiency (CYP19Α1: P450arom)
Aromatase (P450arom) catalyses the conversion of androgens (C19 steroids) to estrogens (C18 steroids), which is the key step in estrogen biosynthesis. P450arom is encoded by the CYP19 gene localized on chromosome 15p21.1 (ΟMIM#107910).81 This enzyme is mainly located in the endoplasmic reticulum of estrogen producing organs such as the ovary, the placenta, breast, and bone.81 Aromatization of fetal adrenal androgens is essential for production of estrogens during pregnancy by the human placenta, the principal products of which are estriol, estrone and estradiol. Therefore, a placental defect in aromatization results in low estrogen production during pregnancy.
Aromatase deficiency is rare and leads to virilization of the mother during pregnancy and exposure of the female fetus to adrenal androgens with consequent ambiguous genitalia, elevated androgens and undetectable estrogens at birth.82-84 At a later stage, aromatase deficiency is associated with lack of breast development, primary amenorrhea, tall stature and multicystic ovaries, developing not only in adolescent girls but also during infancy and childhood.82,84 It has been proposed that multicystic ovaries are secondary to high serum gonadotrophins concentrations and low serum estradiol levels, which are required to restrain FSH and LH secretion.82 Aromatase mutations can produce variable or “nonclassic” female phenotypes including genital ambiguity at birth, but with variable breast development at puberty (B2–B4), and absent puberty with minimal androgenization at birth, suggesting that low residual aromatase activity may be sufficient for breast and uterine development at puberty despite significant androgenization in utero.84 Furthermore, aromatase insufficiency may have wider implications in the general female population, as polymorphic variability within the aromatase locus has been associated with hyperandrogenism in younger women and variations in bone mineral density and fracture risk in postmenopausal women.85,86
3. Maternal (luteoma, exogenous)
In the past, some of the synthetic progestins prescribed to women with recurrent miscarriages exerted some androgenic activity with concomitant virilization of the external genitalia of the female fetuses. Today such progestins are no longer prescribed and modern progesterone preparations do not have such strong androgenic effects, therefore cases of iatrogenic virilization of the external genitalia of the female fetuses are now extremely rare.
Masculinization of the external genitalia of female fetuses has been described in pregnancies of mothers harboring a virilizing adrenocortical, ovarian or Krukenberg (an ovarian metastasis of a primary tumor derived from abdominal or retroperitoneal organs) tumor or a luteoma of pregnancy.87-89 Luteoma of pregnancy is a non-neoplastic hormone-dependent tumor-like lesion of the ovary that occurs during pregnancy and regresses after delivery. Although luteomas are usually asymptomatic, in 25% of cases they are hormonally active, with secretion of androgens, resulting in masculinization of mothers and virilization of the external genitalia of female infants of varying degrees.89
Untreated CAH of the mother may lead to virilization of the external genitalia of the female infant; however, careful management and monitoring leads to normal female infants with nonvirilized external genitalia, which points to the importance of good control during pregnancy of women with CAH.90,91
C. Other abnormalities [cloacal exstrophy, vaginal atresia, MURCS (Müllerian, renal, cervicothoracic somite abnormalities), other syndromes]
This category includes anomalies such as vaginal atresia, cloacal exstrophy, uterine anomalies, (M?llerian agenesis/hypoplasia), labial adhesions.
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome (ΟΜΙΜ#277000). This syndrome is characterized by aplasia of the uterus and the upper part of the vagina in an individual with a normal 46,XX karyotype, normal ovarian function and hence normal development of secondary sexual characteristics during puberty.92 The incidence of the MRKH syndrome has been estimated at 1 in 4,500 female births. The majority of cases are sporadic; however, familial cases have also been described. The first clinical manifestation is primary amenorrhea in patients presenting with normal function of the ovaries and no signs of androgen excess. Isolated utero-vaginal aplasia with normal Fallopian tubes is referred to as Rokitansky sequence or type I (isolated) MRKH syndrome. Incomplete uterine aplasia with hypoplasia or aplasia of one or the two tubes and/or other malformations is generally referred to as MURCS association or type II MRKH syndrome (ΟΜΙΜ# 601076). Type II or MURCS is associated with renal (unilateral renal agenesis, ectopia of kidneys or horseshoe kidney), skeletal and, in particular, vertebral anomalies, hearing defects and, more rarely, cardiac and digital anomalies. Although several genes with a broad spectrum of activity during early development (such as WT1, PAX2, HOXA7 to HOXA13 and PBX1) have been suggested as candidates, the etiology of type II MRKH syndrome remains unknown. WNT4 mutations in XX women lead to a syndrome characterized by absence of Müllerian duct derivatives and hyperandrogenism with or without renal anomalies, which is close to but is considered different from MRKH syndrome.26
CONCLUSIONS
The new nomenclature, as proposed by the consensus statement, has resulted in a more etiologically precise classification system, which has the significant advantage of incorporating both the karyotype and molecular abnormalities. Of some concern is the fact that, although socially stigmatising terms are largely avoided, the new terminology still carries the term ‘sex disorders’, which may be considered pejorative by the patients.4 Nevertheless, while the new terminology may not be perfect, it provides more clarity, thus enhancing understanding of DSD and their management, and is gradually being accepted and included in standard endocrine textbooks.93
There is still much to learn about DSD disorders, this evidenced by the fact that, though the process of ovarian development is under intense investigation, many aspects of sex determination and differentiation in the 46,XX individual remain unknown. DSD disorders in the 46,XX individual are rare, the commonest being CAH. Impressive advances in the field of genetics are producing an expanding list of disorders in the 46,XX individual, as briefly discussed in this review, which now includes very rare disorders such as aromatase deficiency and POR deficiency. While rapid progress in disclosing impaired pathways has been achieved, much more research is needed for the full understanding of pathogenetic mechanisms, which will lead to improvement in diagnostic procedures and management.
REFERENCES
1. MacLaughlin DT, Donahoe PK, 2004 Sex determination and differentiation. N Engl J Med 350: 367-378.
2. Hughes IA, 2004 Female Development — All by Default? N Engl J Med 351: 748-750.
3. Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES Consensus Group, ESPE Consensus Group, 2006 Consensus statement on management of intersex disorders. Arch Dis Child 91: 554-563.
4. Dacou-Voutetakis C, 2007 A multidisciplinary approach to the management of children with complex genital anomalies. Nat Clin Pract Endocrinol Metab 3: 668-669.
5. Sadler TW, 2006 Third to eight weeks: The embryonic period. In: Lippincott Williams & Wilkins, (eds) Langman’s Medical Embryology 10th edition, Baltimore, Maryland, USA; pp, 67-87.
6. Sadler TW, 2006 Urogenital system. In: Lippincott Williams & Wilkins (eds) Langman’s Medical Embryology 10th edition, Baltimore, Maryland, USA; pp, 229-256.
7. Sadler TW, 2006 Gametogenesis: Conversion of germ cells into male and female gametes. In: Lippincott Williams & Wilkins (eds) Langman’s Medical Embryology 10th edition, Baltimore, Maryland, USA; pp, 11-29.
8. Wilhelm D, Palmer S, Koopman P, 2007 Sex determination and gonadal development in mammals. Physiol Rev 87: 1-28.
9. Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al, 1999 The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms’ tumor. Hum Mutat 13: 146-153.
10. Jaubert F, Vasiliu V, Patey-Mariaud de Serre N, et al, 2003 Gonad development in Drash and Frasier syndromes depends on WT1 mutations. Arkh Patol 65: 40-44.
11. Demmer L, Primack W, Loik V, Brown R, Therville N, McElreavey K, 1999 Frasier syndrome: a cause of focal segmental glomerulosclerosis in a 46,XX female. J Am Soc Nephrol 10: 2215-2218.
12. Parker KL, Rice DA, Lala DS, et al, 2002 Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res 57: 19-36.
13. Lala DS, Rice DA, Parker KL, 1992 Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu-factor I. Mol Endocrinol 6: 1249-1258.
14. Mallet D, Bretones P, Michel-Calemard L, Dijoud F, David M, Morel Y, 2004 Gonadal dysgenesis without adrenal insufficiency in a 46,XY patient heterozygous for the nonsense C16X mutation: a case of SF1 haploinsufficiency. J Clin Endocrinol Metab 89: 4829-4832.
15. Achermann JC, Ito M, Ito M, Hindmarsh PC, Jameson JL, 1999 A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet 22: 125-126.
16. Philibert P, Zenaty D, Lin L, et al, 2007 Mutational analysis of steroidogenic factor 1 (NR5a1) in 24 boys with bilateral anorchia: a French collaborative study. Hum Reprod 22: 3255-3261.
17. K?hler B, Lin L, Ferraz-de-Souza B, et al, 2008 Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum Mutat 29: 59-64.
18. Lin L, Achermann JC, 2008 Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev 2: 200-209.
19. Biason-Lauber A, Schoenle EJ, 2000 Apparently normal ovarian differentiation in a prepubertal girl with transcriptionally inactive steroidogenic factor 1 (NR5A1/SF-1) and adrenocortical insufficiency. Am J Hum Genet 67: 1563-1568.
20. Jeays-Ward K, Hoyle C, Brennan J, et al, 2003 Endothelial and steroidogenic cell migration are regulated by WNT4 in the developing mammalian gonad. Development 130: 3663-3670.
21. Yao HH, Matzuk MM, Jorgez CJ, et al, 2004 Follistatin operates downstream of Wnt4 in mammalian ovary organogenesis. Dev Dyn 230: 210-215.
22. Heikkil? M, Prunskaite R, Naillat F, et al, 2005 The partial female to male sex reversal in Wnt-4-deficient females involves induced expression of testosterone biosynthetic genes and testosterone production, and depends on androgen action. Endocrinology 146: 4016-4023.
23. Vainio S, Heikkil? M, Kispert A, Chin N, McMahon AP, 1999 Female development in mammals is regulated by Wnt-4 signalling. Nature 397: 405-409.
24. Philibert P, Biason-Lauber A, Rouzier R, et al, 2008 Identification and functional analysis of a new WNT4 gene mutation among 28 adolescent girls with primary amenorrhea and M?llerian duct abnormalities: a French collaborative study. J Clin Endocrinol Metab 93: 895-900.
25. Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ, 2004 A WNT4 mutation associated with M?llerian-duct regression and virilization in a 46,XX woman. N Engl J Med 351: 792-798.
26. Biason-Lauber A, De Filippo G, Konrad D, Scarano G, Nazzaro A, Schoenle EJ, 2007 WNT4 deficiency-a clinical phenotype distinct from the classic Mayer-Rokitansky-K?ster-Hauser syndrome: a case report. Hum Reprod 22: 224-229.
27. Zanaria E, Muscatelli F, Bardoni B, et al, 1994 An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature 372: 635-641.
28. Bardoni B, Zanaria E, Guioli S, et al, 1994 A dosage sensitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat Genet 7: 497-501.
29. Seminara SB, Achermann JC, Genel M, Jameson JL, Crowley WF, Jr, 1999 X-linked adrenal hypoplasia congenita: a mutation in DAX1 expands the phenotypic spectrum in males and females. J Clin Endocrinol Metab 84: 4501-4509.
30. Yu RN, Ito M, Saunders TL, Camper SA, Jameson JL, 1998 Role of Ahch in gonadal development and gametogenesis. Nat Genet 20: 353-357.
31. Mizusaki H, Kawabe K, Mukai T, et al, 2003 Dax-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1) gene transcription is regulated by wnt4 in the female developing gonad. Mol Endocrinol 17: 507-519.
32. Schmidt D, Ovitt CE, Anlag K, et al, 2004 The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development 131: 933-942.
33. Pailhoux E, Vigier B, Vaiman D, et al, 2002 Ontogenesis of female-to-male sex-reversal in XX polled goats. Dev Dyn 224: 39-50.
34. Beysen D, Vandesompele J, Messiaen L, De Paepe A, De Baere E, 2004 The human FOXL2 mutation database. Hum Mutat 24: 189-193.
35. Crisponi L, Deiana M, Loi A, et al, 2001 The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet 27: 159-166
36. Uhlenhaut NH, Jakob S, Anlag K, et al, 2009 Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 139: 1130-1142.
37. Kim KA, Zhao J, Andarmani S, et al, 2006 R-Spondin proteins: a novel link to beta-catenin activation. Cell Cycle 5: 23-26.
38. Tomizuka K, Horikoshi K, Kitada R, et al, 2008 R-spondin1 plays an essential role in ovarian development through positively regulating Wnt-4 signaling. Hum Mol Genet 17: 1278-1291.
39. Kocer A, Pinheiro I, Pannetier M, et al, 2008 R-spondin1 and FOXL2 act into two distinct cellular types during goat ovarian differentiation. BMC Dev Biol 8: 36.
40. Parma P, Radi O, Vidal V, et al, 2006 R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet 38: 1304-1309.
41. Tomaselli S, Megiorni F, De Bernardo C, et al, 2008 Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat 29: 220-226.
42. Krob G, Braun A, Kuhnle U, 1994 True hermaphroditism: geographical distribution, clinical findings, chromosomes and gonadal histology. Eur J Pediatr 153: 2-10.
43. Verkauskas G, Jaubert F, Lortat-Jacob S, Malan V, Thibaud E, Nihoul-F?k?t? C, 2007 The long-term follow up of 33 cases of true hermaphroditism: a 40-year experience with conservative gonadal surgery. J Urol 177: 726-731.
44. Hadjiathanasiou CG, Brauner R, Lortat-Jacob S, et al, 1994 True hermaphroditism: genetic variants and clinical management. J Pediatr 125: 738-744.
45. Damiani D, Fellous M, McElreavey K, et al, 1997 True hermaphroditism: clinical aspects and molecular studies in 16 cases. Eur J Endocrinol 136: 201-204.
46. Berkovitz GD, Fechner PY, Marcantonio SM, et al, 1992 The role of the sex-determining region of the Y chromosome (SRY) in the etiology of 46,XX true hermaphroditism. Hum Genet 88: 411-416.
47. Ortenberg J, Oddoux C, Craver R, et al, 2002 SRY gene expression in the ovotestes of XX true hermaphrodites. J Urol 167:1828-1831.
48. de la Chapelle A, 1981 The etiology of maleness in XX men. Hum Genet 58: 105-116.
49. L?pez M, Torres L, M?ndez JP, et al, 1995 Clinical traits and molecular findings in 46,XX males. Clin Genet 48: 29-34.
50. Vorona E, Zitzmann M, Gromoll J, Sch?ring AN, Nieschlag E, 2007 Clinical, endocrinological and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J Clin Endocrinol Metab 92: 3458-3465.
51. Abbas N, McElreavey K, Leconiat M, et al, 1993 Familial case of 46,XX male and 46,XX true hermaphrodite associated with a paternal-derived SRY-bearing X chromosome. C R Acad Sci III 316: 375-383.
52. Ergun-Longmire B, Vinci G, Alonso L, et al, 2005 Clinical, hormonal and cytogenetic evaluation of 46,XX males and review of the literature. J Pediatr Endocrinol Metab 18: 739-748.
53. Huang B, Wang S, Ning Y, Lamb AN, Bartley J, 1999 Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet 87: 349-353.
54. Seeherunvong T, Perera EM, Bao Y, et al, 2004 46,XX sex reversal with partial duplication of chromosome arm 22q. Am J Med Genet A 127: 149-151.
55. Skordis NA, Stetka DG, MacGillivray MH, Greenfield SP, 1987 Familial 46,XX males coexisting with familial 46,XX true hermaphrodites in same pedigree. J Pediatr 110: 244-248
56. Sarafoglou K, Ostrer H, 2000 Clinical review 111: familial sex reversal: a review. J Clin Endocrinol Metab 85: 483-493.
57. Gore CA, 2010 Neuroendocrine targets of endocrine disruptors. Hormones (Athens) 9: 16-27.
58. Migeon CJ, Wisniewski AB, 2003 Human sex differentiation and its abnormalities. Human sex differentiation and its abnormalities. Best Pract Res Clin Obstet Gynaecol 17: 1-18.
59. Meyers CM, Boughman JA, Rivas M, Wilroy RS, Simpson JL, 1996 Gonadal (ovarian) dysgenesis in 46,XX individuals: frequency of the autosomal recessive form. Am J Med Genet 63: 518-524.
60. Pallister PD, Opitz JM, 1979 The Perrault syndrome: autosomal recessive ovarian dysgenesis with facultative, non-sex-limited sensorineural deafness. Am J Med Genet 4: 239-246.
61. Nishi Y, Hamamoto K, Kajiyama M, Kawamura I, 1988 The Perrault syndrome: clinical report and review. Am J Med Genet: 31: 623-629.
62. Aittom?ki K, Herva R, Stenman UH, et al, 1996 Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene. J Clin Endocrinol Metab 81: 3722-3776.
63. Marozzi A, Manfredini E, Tibiletti MG, et al 2000 Molecular definition of Xq common-deleted region in patients affected by premature ovarian failure. Hum Genet 107: 304-311.
64. Rossetti R, Di Pasquale E, Marozzi A, et al, 2009 BMP15 mutations associated with primary ovarian insufficiency cause a defective production of bioactive protein. Hum Mutat 30: 804-810.
65. Murray A, Webb J, Grimley S, Conway G, Jacobs P, 1998 Studies of FRAXA and FRAXE in women with premature ovarian failure. J Med Genet 35: 637-640.
66. Massad-Costa AM, da Silva ID, Affonso R, et al, 2007 Gene analysis in patients with premature ovarian failure or gonadal dysgenesis: a preliminary study. Maturitas 57: 399-404.
67. Holland C, 2000 47,XXX in an adolescent with premature ovarian failure and autoimmune disease. J Pediatr Adolesc Gynecol 13: 93.
68. Schlessinger D, Herrera L, Crisponi L, Mumm S, Percesepe A, Pellegrini M, 2002 Genes and translocations involved in POF. Am J Med Genet 111: 328-333.
69. Dacou-Voutetakis C, Maniati-Christidi M, Dracopoulou-Vabouli M, 2001 Genetic aspects of congenital adrenal hyperplasia. J Pediatr Endocrinol Metab 14: Suppl 5: 1303-1308.
70. Krone N, Dhir V, Ivison HE, Arlt W, 2007 Congenital adrenal hyperplasia and P450 oxidoreductase deficiency. Clin Endocrinol (Oxf) 66: 162-172.
71. Merke DP, Bornstein SR, 2005 Congenital adrenal hyperplasia. Lancet 365: 2125-2136.
72. Tonetto-Fernandes V, Lemos-Marini SH, Kuperman H, Ribeiro-Neto LM, Verreschi IT, Kater CE, 2006 Serum 21-Deoxycortisol, 17-Hydroxyprogesterone, and 11-deoxycortisol in classic congenital adrenal hyperplasia: clinical and hormonal correlations and identification of patients with 11beta-hydroxylase deficiency among a large group with alleged 21-hydroxylase deficiency. J Clin Endocrinol Metab 91: 2179-2184.
73. Moran C, Azziz R, Carmina E, et al, 2000 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Am J Obstet Gynecol 183: 1468-1474.
74. Dacou-Voutetakis C, Dracopoulou M, 1999 High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab 84: 1570-1574.
75. Dracopoulou-Vabouli M, Maniati-Christidi M, Dacou-Voutetakis C, 2001 The spectrum of molecular defects of the CYP21 gene in the Hellenic population: variable concordance between genotype and phenotype in the different forms of congenital adrenal hyperplasia. J Clin Endocrinol Metab 86: 2845-2848.
76. Joehrer K, Geley S, Strasser-Wozak EM, et al, 1997 CYP11B1 mutations causing non-classic adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Hum Mol Genet 6: 1829-1834.
77. Scott RR, Miller WL, 2008 Genetic and Clinical Features of P450 Oxidoreductase Deficiency. Horm Res 69: 266-275.
78. Arlt W, Walker EA, Draper N, et al, 2004 Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 363: 2128-2135.
79. Mendonca BB, Leite MV, de Castro M, et al, 2002 Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab 87: 1805-1809.
80. Charmandari E, Kino T, Ichijo T, Chrousos GP, 2008 Generalized Glucocorticoid Resistance: Clinical Aspects, Molecular Mechanisms and Implications of a Rare Genetic Disorder. J Clin Endocrinol Metab 93: 1563-1572.
81. Payne AH, Hales DB, 2004 Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev 25: 947-970.
82. Mullis PE, Yoshimura N, Kuhlmann B, Lippuner K, Jaeger P, Harada H, 1997 Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab 82: 1739-1745.
83. Belgorosky A, Pepe C, Marino R, et al, 2003 Hypothalamic-pituitary-ovarian axis during infancy, early and late prepuberty in an aromatase-deficient girl who is a compound heterozygote for two new point mutations of the CYP19 gene. J Clin Endocrinol Metab 88: 5127-5131.
84. Lin L, Ercan O, Raza J, et al, 2007 Variable phenotypes associated with aromatase (CYP19) insufficiency in humans. J Clin Endocrinol Metab 92: 982-990.
85. Masi L, Becherini L, Gennari L, et al, 2001 Polymorphism of the aromatase gene in postmenopausal Italian women: distribution and correlation with bone mass and fracture risk. J Clin Endocrinol Metab 86: 2263-2269.
86. Petry CJ, Ong KK, Michelmore KF, et al, 2005 Association of aromatase (CYP 19) gene variation with features of hyperandrogenism in two populations of young women. Hum Reprod 20: 1837-1843.
87. Kirk JMW, Perry LA, Shand WS, Kirby RS, Besser GM, Savage MO, 1990 Female pseudohermaphroditism due to a maternal adrenocortical tumor. J Clin Endocrinol Metab 70: 1280-1284.
88. Vauthier-Brouzes D, Vanna Lim-You K, Sebagh E, Lefebvre G, Darbois Y, 1997 Krukenberg tumor during pregnancy with maternal and fetal virilization: a difficult diagnosis. A case report. J Gynecol Obstet Biol Reprod (Paris) 26: 831-833.
89. Mazza V, Di Monte I, Ceccarelli PL, et al, 2002 Prenatal diagnosis of female pseudohermaphroditism associated with bilateral luteoma of pregnancy: case report. Hum Reprod 17: 821-824.
90. Kai H, Nose O, Iida Y, Ono J, Harada T, Yabuuchi H, 1979 Female pseudohermaphroditism caused by maternal congenital adrenal hyperplasia. J Pediatr 95: 418-420.
91. Lo JC, Schwitzgebel VM, Tyrrell JB, et al, 1999 Normal female infants born of mothers with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 84: 930-936.
92. Morcel K, Camborieux L; Programme de Recherches sur les Aplasies M?llιriennes, Guerrier D, 2007 Mayer-Rokitansky-K?ster-Hauser (MRKH) syndrome. Orphanet J Rare Dis 2: 13.
93. Achermann JC, Hughes IA, 2008 Disorders of sex development. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR (eds) Williams Textbook of Endocrinology 11th edition, Philadelphia; pp, 783-848.
Address for correspondence:
Eleni Kousta, 6 S. Arvanitaki street, Corfu 49100, Greece,
Τel: + 30 26610 80561, Fax: +30 26610 80562, Mob: +30 697 482816, e-mail: lkousta@otenet.gr
Received 08-03-10, Revised 10-05-10, Accepted 30-05-10