Center for Molecular and Cellular Toxicology, Division of Pharmacology & Toxicology, College of Pharmacy; Institute for Cellular and Molecular Biology; and Institute for Neuroscience, the University of Texas, Austin, U.S.A.
The central neuroendocrine systems are responsible for the control of homeostatic processes in the body, including reproduction, growth, metabolism and energy balance, as well as stress responsiveness. These processes are initiated by signals in the central nervous system, specifically the hypothalamus, and are conveyed first by neural and then by endocrine effectors. The neuroendocrine systems, as the links between the brain and peripheral endocrine organs, play critical roles in the ability of an organism to respond to its environment under normal circumstances. When neuroendocrine homeostasis is disrupted by environmental endocrine-disrupting chemicals, a variety of perturbations can ensue, particularly when endocrine disruption occurs during critical developmental time periods. This article will discuss the evidence for environmental endocrine disruption of neuroendocrine systems and the effects on endocrine and reproductive functions.
Endocrine disruption, Epigenetic, Fetal basis of adult disease, GnRH, Hypothalamus, Sex differences, Thyroid, Transgenerational
1. OVERVIEW OF NEUROENDOCRINE SYSTEMS
The hypothalamus, located at the base of the brain, is the site of the neurons that control central neuroendocrine function in vertebrates. The neuroendocrine hypothalamus, which serves as a major interface between the central nervous system and the rest of the body, signals to the periphery through the release of hypothalamic releasing/inhibiting hormones into the portal capillary system that leads to the anterior pituitary gland (Figure 1). Specifically, six classes of hypothalamic neural cells regulate the anterior pituitary gland and they are named for the hormones that they produce (summarized in Table 1 ): corticotropin-releasing hormone (CRH); gonadotropin-releasing hormone (GnRH); growth hormone-releasing hormone (GHRH) and somatostatin; thyrotropin-releasing hormone (TRH); and dopamine (prolactin-inhibiting hormone). These neurotransmitters are synthesized in hypothalamic neurons, packaged into secretory vesicles, and released directly into the portal capillary system that vascularizes the anterior pituitary. The pituitary in turn releases its corresponding hormones (Table 1 ). Finally, the peripheral endocrine organs release hormones into the bloodstream that exert a crucial homeostatic regulatory function. The target hormones also feed back to neuroendocrine cells in the hypothalamus to communicate information about whether more or less hormonal production is necessary to maintain homeostasis.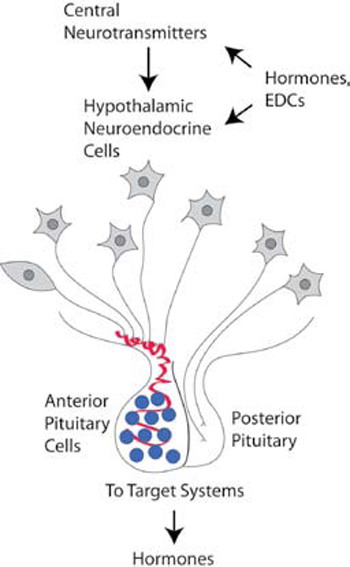
Figure 1. Schematic depiction of how hypothalamic neuroendocrine systems are targets of environmental endocrine disruptors. The hypothalamic neurons are depicted as neurons with axons terminating in the portal capillary vasculature (red) that leads to the anterior pituitary, or directly to the posterior pituitary. From the pituitary gland, additional hormones are released that act upon target cells elsewhere in the body, many of which secrete another level of hormones. In the brain, hypothalamic neuroendocrine cells are regulated by neurotransmitter systems and by both direct and indirect hormone actions. Environmental endocrine-disrupting chemicals (EDCs) may mimic or block some of these hormonal effects in the brain, thereby disrupting neuroendocrine processes.
There is increasing evidence that the central neuroendocrine systems are targets of endocrine-disrupting chemicals (EDCs). For the purposes of this article, I define endocrine disruptors as those compounds that interfere with hormonally regulated physiological processes. They include industrial compounds, plastics and plasticizers, fungicides, pesticides, and others. In some cases, pharmaceuticals and phytoestrogens are deemed EDCs when consumed in large amounts (e.g. soy supplements), or when pharmaceuticals are given at high levels at inappropriate developmental periods (e.g. pregnancy), or when they pass into the environment through urination. Because many endocrine disruptors are environmental contaminants from water, food, air and soil, I also use the terminology of “environmental endocrine disruptors” throughout. The mechanisms of EDCs may vary (e.g. binding to nuclear hormone receptors to stimulate or antagonize them; interference with enzymes involved in steroid biosynthesis; neurotransmitter systems that are steroid sensitive; hormone degradation/elimination) but the net effect on an organism is an altered endocrine phenotype. With respect to neuroendocrine systems, as they both control and respond to hormonal signaling, they are logical targets for endocrine disruption. Further, neuroendocrine cells are neurons and therefore they are capable of responding to external stimuli such as environmental signals very rapidly, in the order of milliseconds to seconds. In addition, because neuroendocrine cells are also endocrine effectors, their responses can be protracted for hours, days, or even weeks. Thus, the hypothalamic neuroendocrine systems are perfectly poised to mediate responses to environmental stimuli, including endocrine disruptors.
2. ENVIRONMENTAL ENDOCRINE DISRUPTION OF NEUROENDOCRINE SYSTEMS
There is increasing interest and research regarding the effects of environmental endocrine disruptors on neuroendocrine systems.1-3 Most studies have focused on the Hypothalamic-Pituitary-Gonadal (HPG) axis, and hence, this article will primarily discuss the literature on that subject. However, other systems are vulnerable to endocrine disruption. There is a body of literature showing that the Hypothalamic-Pituitary-Thyroid (HPT) axis, is highly susceptible to endocrine disruption. Relatively few studies have investigated links between endocrine disruptors and stress [Hypothalamic-Pituitary-Adrenal (HPA) axis], growth, or lactation, and therefore such data will not be presented in the current article.
The diverse structures and properties of EDCs cause them to exert diverse actions upon target cells. Some EDCs are agonists of hormone receptors expressed on neuroendocrine cells. For example, estrogen receptors can bind EDCs, including Polychlorinated Biphenyls (PCBs), phytoestrogens, pesticides, compounds in plastics such as bisphenol A, and other chemicals.4-6 Dioxins and some PCBs are potent agonists to the aryl hydrocarbon receptor (AhR),7 which is abundantly expressed in the brain.8 Other EDCs may act as antagonists to hormone receptors or as mixed agonist/antagonists. For example, PCBs can cause activation or suppression of thyroid hormone receptor activity depending upon the specific PCB mixture or dose. Phthalates are androgen receptor antagonists, and vinclozolin, a fungicide, acts, at least in part, as an anti-androgen.
Although not the subject of this review, it is important to mention that EDCs may exert their actions upon neurotransmitter systems that regulate neuroendocrine cells. PCBs have been shown to bind to serotonin, dopamine, and noradrenergic receptors.9 As these neurotransmitters act upon the hypothalamic releasing factors that control the pituitary gland, this is another mechanism for neuroendocrine disruption.
3. DEVELOPMENTAL EXPOSURE TO NEUROENDOCRINE DISRUPTORS
The timing of exposure to an EDC is crucial in determining its ultimate effect. It is recognized that there are critical developmental periods during which neuroendocrine systems are modulated by steroid and other hormones. For example, early life exposure to endogenous androgens or estrogens, particularly in fetal life and infancy, organizes the brain in a sexually dimorphic manner (i.e. resulting in morphological and functional differences between males and females) that becomes activated later in life.10-12 Exposure to exogenous substances such as EDCs is likely to have more profound detrimental consequences in developing organisms than in adults.12,13 This concept is now referred to as the “fetal/developmental basis of adult disease” and is highly applicable to neuroendocrine systems. For example, in the case of the HPG axis, early life exposures to environmental EDCs can permanently alter sexual development, resulting in females that are masculinized or defeminized and males that are feminized or demasculinized.14-17 As discussed below, these effects of EDCs on brain sexual differentiation are manifested as changes in reproductive development and may be detrimental to fertility and reproductive success. Therefore, the fetal/developmental basis of adult disease is a critical concept for neuroendocrine disruption.
4. ENDOCRINE DISRUPTION OF SEXUAL DIFFERENTIATION OF THE BRAIN
Neuroendocrinologists have known for decades that early life exposure to hormones such as estradiol or testosterone can permanently alter the morphology of the brain, together with the development of sex-typical reproductive physiology and behavior.10-12,18-21 However, the mechanisms for these effects of hormones on the brain are still poorly understood and quite controversial, partially on account of the complexity of these systems. Studies on normal development, and on effects of endogenous hormones in these processes, make it clear that sexual differentiation of the brain results in large part from the actions of fetal and maternal hormones on steroid hormone receptors in the brain.10-12,18-21 Sexual differentiation is also regulated by metabolism of steroid hormones in the brain by the enzymes controlling steroidogenesis, such as p450 aromatase, which converts testosterone to estradiol.22,23 Further sex differences are due to circulating alpha-fetoprotein, a binding protein that protects the fetal brain from exposure to circulating maternal estrogens.24 In rodents and many other mammals, the male fetal gonad produces high levels of testosterone; this hormone can serve as a precursor of estradiol through its aromatization within neural tissues of males.21 By contrast, the female fetal gonad produces much lower levels of hormones and the female brain is “protected” from maternal estradiol by alpha-fetoprotein, as shown in mouse models.21 Developmental apoptosis (programmed cell death) in hypothalamic nuclei also plays a role in sexual differentiation, a process that is profoundly influenced by the steroid hormonal environment.25-27 Any and all of these mechanisms can be disrupted by environmental compounds, particularly when exposure occurs during the developmental periods when these processes are most active, namely, embryonic and early postnatal development.
Clearly, the mechanisms for brain sexual differentiation are complex. Seeking to understand endocrine disruption of brain sexual differentiation is quite difficult, but emerging data support effects of EDC exposure on the development of sexually dimorphic circuits in the brain, with functional consequences (reviewed in reference 2). Studies on environmental endocrine disruption of hypothalamic morphology show effects of developmental exposures. It is important to point out, though, that the effects tend to be specific to certain cellular phenotypes rather than to the size/volume of a region as a whole. For example, early postnatal injections of rat pups (during the first two days of life) with the estrogenic endocrine disruptor bisphenol A, or the phytoestrogen genistein, did not affect the total volume of the sexually dimorphic nucleus of the hypothalamus (SDN), but increased the number of those cells immunoreactive for the calcium binding protein, calbindin, in this region of male rats.28 The SDN is a dimorphic nucleus that normally is larger in male than in female rats;29 thus, perinatal endocrine disruptor exposure hyper-masculinized calbindin cell numbers in this region of the males without affecting overall regional volume. In that same study28 in the anteroventral periventricular nucleus (AVPV), a brain region important for the neuroendocrine control of ovulation in females and potentially involved in neuroendocrine control of GnRH release in both sexes,30 genistein increased the AVPV volume of the males. This shows that in a subset of experiments, the size of a region can be altered by neonatal endocrine disruptors.
Neuroendocrine systems also control behaviors, and there are several studies showing disruption of reproductive behaviors in male and female animal models. Low doses of PCBs given to developing rats31,32 or soy phytoestrogens given to female rats15 caused quantifiable adverse changes in reproductive behavior. A study published by my laboratory showed that treatment of a pregnant rat on days 16 and 18 of gestation with low levels of the PCB mixture, Aroclor 1221, affected the mating behavior of her female offspring in adulthood. This fetally exposed F1 generation showed a decrease in the likelihood of engaging in mating behavior, and such F1 females, when they did mate, spent more time away from the male in a paced mating trial.32 Chlordecone, an estrogenic pesticide given at a single embryonic age to rats (day 16 of gestation), permanently altered sexual behavior in adulthood.33 In that study, the adult female F1 rats showed an enhanced lordosis quotient, an index of receptivity. Male rats showed an enhanced lordosis rating (intensity of the lordosis posture), suggesting that they were feminized by the prenatal treatment with chlordecone. Both males and females also showed enhancements of mounting behavior, suggestive of a hypermasculinization of the males and a masculinization of the females. The phytoestrogen, coumestrol, given during early postnatal life caused a reduction in masculine34 and feminine sexual behaviors.35 A recent study evaluated effects of a proposed endocrine disruptor found in ultraviolet filters, 4-methylbenzylidene camphor (4-MBC), on sexual behaviors in female rats. Exposure to 4-MBC throughout the life cycle caused a diminution in proceptive behaviors and affected the females apparent attractiveness to normal (untreated) males. Lordosis quotient and receptivity were also diminished.36
As a whole, these data show that EDC exposures during development cause aberrant reproductive behavioral outcomes in adulthood. If an animal is not behaving in a stereotypical way, or if behaviors are compromised, the animal may be less likely to reproduce, but even if he/she has a residual capacity to mate, the individual may not be selected by a conspecific who can choose an unaffected mate over an affected one. Thus, there are evolutionary and ecological consequences to the effects of EDCs on reproductive behaviors.
5. NEUROENDOCRINE DISRUPTION OF THE HPG AXIS
The hypothalamic GnRH neurons control reproductive function in all vertebrates.37 These cells are named for the decapeptide synthesized in their cell bodies. Only about 800 neurons of mammalian brains are capable of producing and releasing GnRH. First, I will provide a brief overview of how GnRH neurons regulate the other levels of the hypothalamic-pituitary-gonadal axis and how GnRH cells in turn are regulated by feedback from peripheral sex steroid hormones. The GnRH neural cells project their axons to the base of the hypothalamus (called the median eminence or infundibulum, depending upon species), where the GnRH decapeptide is released into the portal capillaries that vascularize the anterior pituitary gland. At the pituitary gland, GnRH binds to its receptors on a subset of cells called gonadotropes, which synthesize and release LH and FSH into the general circulation (Table 1 ). LH and FSH bind to receptors on the ovary or the testis to cause steroidogenesis and gametogenesis. The gonadal steroid hormones that are produced act upon reproductive and other targets in the body and brain, including feedback regulation of the GnRH neurons. In most mammals, including humans, non-human primates and rodents, 17β-estradiol, testosterone, and progesterone are the major estrogens, androgens, and progestins, respectively, and their receptors are abundantly expressed in the hypothalamus.38,39
There is in vitro and in vivo evidence that GnRH neurons can be direct targets of EDCs. The GnRH GT1-7 cell line, an in vitro immortalized hypothalamic cell line that has many properties of the GnRH cell in vivo, has been used for exploring the molecular mechanisms by which different classes of EDCs may act directly upon GnRH cells. When GT1-7 cells were treated with a PCB mixture, Aroclor 1221 or Aroclor 1254, these cells had elevated GnRH gene expression from low dose treatment and there was relatively little effect by higher doses.40 In that same study, there was a stimulation of GnRH peptide release into the medium by Aroclor 1221 but not Aroclor 1254.40 Further, a nuclear estrogen receptor antagonist, ICI 182,780, blocked some of these effects of PCBs, suggesting a mechanism that is partially mediated by this receptor. In interpreting these data, it is necessary to point out that GT1 cells express the nuclear estrogen receptors, ERβ and ERβ,41,42 but GnRH neurons in the mammalian brain express only ERβ.43,44 Therefore, these reported effects in the GT1-7 cell line may differ somewhat from those in vivo. Finally, Aroclor 1221 caused morphological changes in the GT1-7 cells such as neurite extension and an increase in cellular confluence, whereas Aroclor 1254 caused some modest neurite retraction and neurotoxicity.40 One also needs to consider that PCBs can bind to neurotransmitter receptors,9 some of which are expressed on GT1-7 cells, so this is a potential mechanism by which PCBs activate GnRH gene expression and release.
In a second study, we treated the GT1-7 cell line with the estrogenic organochlorine pesticides, methoxy¬chlor, or chlorpyrifos. These substances significantly increased GnRH gene expression at low doses and inhibited gene expression at high dosages.45 Such an inverted U-shaped dose-response curve is sometimes observed in EDCs effects.46 Although there are many possible explanations for non-linear dose-response curves, I favor the interpretation that multiple pathways are induced by EDCs, each with differing dose-response curves, which when summated result in a non-linear curve. In addition, GT1-7 cell morphology was unaffected by methoxychlor, but chlorpyrifos stimulated neurite outgrowth and cell confluency in a manner similar to estradiol. Overall, these results are important because they show that GnRH cell lines can respond directly to environmental EDCs.
A recent publication from my laboratory has added more information about the mechanisms by which EDCs affect GnRH neurons. GT1-7 cells were treated with either individual PCBs (PCB 74, 118 or 153) or a mixture of the three. Effects on GnRH peptide levels, cell viability and death, and the mechanism for cell death (apoptosis or necrosis) were quantified.47 At low dosages and short time points, GnRH peptide levels were higher than controls. With higher dosages and longer treatment durations, GnRH peptide concentrations were decreased. In general, cellular viability was diminished following PCB treatments in a manner that involved both apoptosis (lower concentrations) and necrosis (higher concentrations).47 These results show that PCBs can impair viability in the GnRH cell line.
In other in vitro work, explanted hypothalamic dissections from 15-day old female rats were incubated in a perifusion system and effects of several EDCs on glutamate-evoked GnRH release were tested (this model was used as a reliable way of stimulating GnRH secretion48). Of the several EDCs tested, o,p’-DDT had the greatest stimulation of glutamate-evoked GnRH release. Bisphenol A also was stimulatory, albeit to a lesser degree. By contrast, the pesticide methoxychlor, and p,p’-DDE, were ineffective in this in vitro model, suggesting some specificity of the effect to certain EDCs.48 The same laboratory showed that DDT, but not DDE, increased the frequency of GnRH pulses, suggesting a stimulatory effect on GnRH release.49 This finding is comparable to the report, discussed above, showing that GnRH release was enhanced by PCBs in the other in vitro model studied, the GT1-7 cells.40 Finally, a recent publication used explanted hypothalami from male rats that had been exposed in utero to dioxin, via treatment of the pregnant dam on gestational day 15.50 That experiment used potassium chloride (KCl)-induced GnRH release in vitro from hypothalami of male rats at day 35, 60, or 90 postnatal, and demonstrated an attenuation of the effects of KCl in the endocrine disrupted males of all three ages.50 At the same time, when GnRH peptide content was measured in the explanted hypothalamus, peptide content was increased, suggesting that the GnRH peptide is synthesized, but release is impaired, in this dioxin model.50 This finding of a diminution in GnRH release contrasts with the studies described earlier showing enhancements of GnRH release in other EDC models. Together, these data make the important point that EDCs can enhance or diminish release of neuroendocrine hormones, depending upon the specific EDC and the timing of treatment, but the net effect is a dysregulated system due to the disruption of homeostasis.
An in vivo animal study supports effects of EDCs on GnRH hypothalamic neurons. In ovariectomized adult rats, McGarvey et al.51 showed that the phytoestrogen coumestrol, but not genistein, suppressed hypothalamic electrical activity together with a suppression of serum LH levels. The hypothalamic activity measured is believed to be representative of the neurophysiological activation of GnRH neurons, reflective of the “GnRH pulse generator” that is responsible for the pulsatile release of GnRH at approximately 30-90 minute intervals. Therefore, a suppression of the GnRH pulse generator suggests that these pulses are not occurring, an interpretation supported by the diminution of LH pulses that would otherwise be stimulated by GnRH pulses.
Studies in the Atlantic croaker fish have shown that PCB treatment can significantly impair HPG and reproductive function.52-54 Specifically, PCBs decrease GnRH peptide content in the hypothalamus, the levels of pituitary GnRH receptors, and the LH response to GnRH challenge.52 Furthermore, the mechanism of action of PCBs in this system appears to be mediated, at least in part, through serotonin receptors, as this neurotransmitter is responsive to PCBs and in turn affects GnRH release.
Developmental exposures to EDCs can disrupt GnRH systems later in life. When female rats were given early postnatal treatments with the phytoestrogen genistein, there was a reduction in the ability of estradiol to induce expression of the immediate early gene Fos within GnRH neurons (a marker of GnRH gene activation). The timing of puberty was altered, and reproductive failure occurred at a much earlier age, in these early postnatally-exposed female rats.1 By contrast, another study by Patisaul et al. showed that expression of the immediate early gene Fos in GnRH neurons was unaffected by neonatal genistein or bisphenol A;28 a reconciliation of these studies requires further research.
Other studies have quantified numbers of GnRH neurons in animals exposed to EDCs during early development. In one, developing rabbits were given prenatal and early postnatal treatment with the anti-androgenic fungicide, vinclozolin.55 A decrease in the number of GnRH neurons was found in specific hypothalamic regions, suggesting a loss of detectable GnRH cells with developmental exposure.55 A recent study used pregnant rats that were treated with TCDD (dioxin) on pregnancy day 15. When their male offsprings were 35 days old (peripubertal), their brains were used for immunohistochemical labeling of GnRH neurons. Prenatal TCDD caused a change in GnRH neural morphology; while it is difficult to know the physiological implications of such a change, it may represent a difference in the capacity for GnRH neurons to receive synaptic inputs.50 Although the results from these studies, one on rabbits, one on rats, may differ, they are consistent in that they suggest that GnRH cells of mammals are targets of EDCs.
6. NEUROENDOCRINE DISRUPTION OF THYROID FUNCTION, METABOLISM, AND ENERGY BALANCE
Although there is extensive research into effects of EDCs on thyroid function (reviewed in reference 56), most research has focused on the thyroid hormones themselves as opposed to the HPT axis. Nevertheless, the HPT axis is sensitive to a broad range of endocrine disruptors, including industrial chemicals (e.g. PCBs, perchlorate), pesticides, and goitrogens in food (reviewed in references 56-58). This is particularly devastating when exposure occurs during development because normal development in mammals and other vertebrates is highly dependent upon a proper hormonal milieu.59 There is some evidence for neuroendocrine HPT disruption: for example, PCB congeners reduce the thyroxine and TSH response to TRH,60 suggesting a hypothalamic and/or pituitary dysregulation. Other studies in rats, humans, and other species provide strong support for thyroid disruption, including pituitary TSH release (reviewed in reference 58).
The importance of the timing of exposure was demonstrated by a study in which pregnant dams were exposed to low doses of polybrominated diphenyl ethers (PBDEs) on day 6 of gestation, resulting in reduced thyroxine levels both in dams and in developing offspring.61 Importantly, developmental exposure to thyroid disruptors can have long-term consequences on neurobiological functions, as thyroid hormone is critical for normal brain development. There is an extensive literature in the case of PCBs, for which it has been reported that neural development is compromised, particularly in the cerebellum, which controls motor learning.62,63 Recently, bisphenol A was identified as a developmental thyroid toxicant, with dosages from 1-50 mg/kg (fed to the dam from gestational day 7 through weaning of the pups) causing transient increases in T4, specifically on postnatal day 15.64 This result highlights the broader nature of endocrine disruption by this compound, formerly thought to be an environmental estrogen.
The control of metabolism and energy balance includes not only euthyroid function but also involves hypothalamic circuits that control feeding behavior, fat cells (adipocytes), and adipokines (e.g. leptin), and actions of the latter on the hypothalamus. Exposure of developing mice to the pharmaceutical estrogenic endocrine disruptor diethylstilbestrol (DES) caused obesity in adulthood, an effect that was manifested in a subsequent generation.65 Although the mechanisms for such effects are not known, it is postulated that it may involve actions of DES on the developing hypothalamic circuits that are important for energy balance. Others have proposed that endocrine disruptors can act as “obesogens” and program the organism to develop obesity later in life through mechanisms including (but probably not limited to) peroxisome proliferator-activated receptors and nuclear hormone receptors involved in lipolysis.56,66 The discipline of the endocrine disruption of metabolism is an emerging field that merits considerably more research. It has implications for obesity, diabetes, and cardiovascular disease, among others, and may be related to the increase in these phenomena in children (reviewed in reference 56).
7. TRANSMISSION OF EFFECTS OF NEUROENDOCRINE DISRUPTION TO FUTURE GENERATIONS
Although it is becoming evident that there can be transgenerational transmission of an endocrine-disrupted trait to future generations65,67,68 and that epigenetic (non-genomic) mechanisms are involved in mediating effects of endocrine disruptors,67,69 this concept has not been broadly applied to neuroendocrine systems. Nevertheless, considering that neuroendocrinologists were among the first to notice that the early life hormonal milieu has permanent organizing effects upon physiology and behavior and development, it seems intuitive that EDC exposure, during development even at sub-toxic levels, will have long-term consequences for the individual when exposures occur during critical ontogenic windows.
The issue of transgenerational transmission of a disrupted trait is suggested by a growing literature concerning the HPA stress neuroendocrine axis, for which early life maternal stress, or even subtle differences in maternal behavior, have been shown to have permanent consequences for stress responsiveness of the offspring and subsequent generations. The mechanism of transmission is also very interesting as it is non-genomic (the transmission of the phenotype is seen in cross-fostered as well as biological offspring). It was determined that this process involved epigenetic modifications to molecules in the brain that were involved in modulating the HPA axis, specifically DNA methylation of the glucocorticoid promoter, and that this trait can be reversed by supplementation with a methyl donor.70,71 This phenomenon of epigenetic programming by maternal behavior has been extended beyond the HPA axis. For example, variations in maternal care are associated with differential methylation of the estrogen receptor alpha promoter.72 Further, the HPG axis is also programmed by early maternal care: rat pups receiving higher levels of licking and grooming have lower indices of sexual behavior, lower responsiveness of GnRH neurons to steroid feedback, and lower expression of ERα in the AVPV.73 It would be very interesting to determine the interactions among endocrine disruptors, maternal behavior, and epigenetic modifications and relate these phenomena to reproductive success.
In terms of EDCs, there are a few models for epigenetic modification and/or transmission of a disrupted trait, and to date, none has addressed neuroendocrine function. However, a summary of that work is presented herein since it may be informative for future neuroendocrine studies. Skinner and colleagues showed that prenatal exposure to the fungicide, vinclozolin, caused disease and dysfunction of the male reproductive system in adulthood, and that this phenotype was transmitted by the male germline for up to at least four generations.67 This research has been replicated and extended by the Anway and Skinner laboratories,67,74 although efforts by other laboratories to replicate this work have had mixed results, possibly due to some experimental differences. Schneider et al75 found that oral administration of vinclozolin to pregnant dams had relatively few reproductive toxicological effects. Their study differed from that of Anway and Skinner in the strain of rat and the route of administration. Inawaka et al also tried to replicate Skinner’s work but did not find a phenotype, possibly due to differences in the rat colonies.76 A recent publication used mice to evaluate transgenerational effects of vinclozolin on DNA methylation in sperm.77 This study is an important extension because not only does it address the question of epigenetic transmission in a new species (mice) but it also focuses on maternally or paternally imprinted genes that are known to be regulated by DNA methylation. Results of treatment of pregnant mouse dams with vinclozolin (compared to vehicle) showed that F1 male sperm had large differences in DNA methylation patterns, with paternally imprinted genes (H19, Gtl2) decreased in percent methylation of CpG sites, and maternally imprinted genes (Peg1, Snrpn, Peg3) increased in percent methylation. Further, these methylation patterns were transmitted to an F2 generation through the F1 male, albeit with a lessening of the effect, and a further diminution in the F3 generation. This loss of the imprinted trait suggests a reversibility of reprogramming of these imprinted genes across generations. The transmission of the trait was not observed with F1 female crosses, supporting a patrilineal mode of inheritance. In the waterflea Daphnia magna, several chemicals were tested, including vinclozolin, for their effects on reproduction and on DNA methylation.78 Although in this experimental invertebrate model vinclozolin did not affect reproduction, it did cause changes in DNA methylation (either hypo- or hypermethylation, depending upon the gene). This effect was also seen in F2 descendants, and in the F3 generation there was global hypomethylation, suggesting transgenerational inheritance of a subset of the epigenetic traits. Unfortunately, the identity of the affected genes is unknown as the Daphnia genome is not yet sequenced, but it will be interesting to compare these results to those in rats and mice, described above, when more information is available. Finally, while not transgenerational, male-specific effects of exposure to vinclozolin (via food) have also been reported for the guppy.79 In that study, feeding female guppies food laced with vinclozolin did not affect their fertility and fecundity, whereas males fed vinclozolin had diminished reproductive success accompanied by decreased sperm counts.79
Two subsequent studies on rats have focused on the neurobiological consequences of transgenerational effects of vinclozolin, bringing research a step closer to understanding links among neuroendocrinology and transgenerational inheritance of non-genomic information. The first evaluated mate choice behavior in F3 descendants of animals exposed to vinclozolin or vehicle and showed that male F3 descendants from the vinclozolin lineage were significantly less attractive to a female mate than those from the vehicle lineage.80 The second study performed microarrays of the transcriptome in whole brain, amygdala, and hippocampus, together with behavioral assays, in the F3 male and female descendants of the two lineages, and found significant alterations in both brain gene expression and behaviors.81 Clearly, more information is needed to understand the relevance of these observations to neuroendocrine systems, but at the very least they show transgenerational transmission of differences in the nervous system in a sex-specific manner.
Using another endocrine disruptor, PCBs, my laboratory published a paper showing effects of prenatal exposure to Aroclor 1221 on physiology and behavior in the adult F1 females and on serum hormones in their adult F2 offspring.32,68 Specific to the F2 generation, we observed that patterns of reproductive hormones on proestrus, the day of the preovulatory GnRH/LH surge, were perturbed.68 In control lineage F2 rats, serum LH and progesterone were high at the predicted time of this surge, whereas in second-generational descendants of PCB-treated pregnant rats, serum LH and progesterone were low. We believe that the timing of the surge may have been disrupted and current research is attempting to address this question, to discern the mechanism for this transmitted trait, and to extend it to an F3 generation.
8. CONCLUSIONS
Central neuroendocrine systems control processes regulated by hypothalamus, pituitary, and target organs. These systems play key roles in enabling the organism to respond to its environment and, thus, they are highly vulnerable to disruption by environmental contaminants such as industrial chemicals, pesticides, plastics/plasticizers, and even food products. To date, most research on endocrine disruption has focused on the target systems, but compelling evidence suggests that the hypothalamic-pituitary levels are potentially the primary sites for endocrine disruption. Further, the neuroendocrine systems do not exist in a vacuum: they communicate with one another and this cross-talk could exacerbate effects of exposures across multiple homeostatic systems.
ACKNOWLEDGMENTS
Work described herein was supported by grants from the NIH (ES018139; ES007247).
REFERENCES
1. Bateman HL, Patisaul HB, 2008 Disrupted female reproductive physiology following neonatal exposure to phytoestrogens or estrogen specific ligands is associated with decreased GnRH activation and kisspeptin fiber density in the hypothalamus. Neurotoxicol 29: 988-997.
2. Dickerson SM, Gore AC, 2007 Estrogenic environmental endocrine-disrupting chemical effects on reproductive neuroendocrine function and dysfunction across the life cycle. Rev Endocrine Metab Disorders 8: 143-159.
3. Gore AC 2007 Endocrine-disrupting chemicals: From basic research to clinical practice. Gore AC (ed) Humana Press; Totowa, NJ.
4. Connor K, Ramamoorthy K, Moore M, et al, 1997 Hydroxylated polychlorinated biphenyls (PCBs) as estrogens and antiestrogens: Structure-activity relationships. Toxicol Appl Pharmacol 145: 111-123.
5. Kuiper GG, Lemmen JG, Carlsson B, et al, 1998 Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139: 4252-4263.
6. Monje L, Varayoud J, Luque EH, Ramos JG, 2007 Neonatal exposure to bisphenol A modifies the abundance of estrogen receptor transcripts with alternative 50-untranslated regions in the female rat preoptic area. J Endocrinol 194: 201-212.
7. Chana A, Concejero MA, de Frutos M, Gonzalez MJ, Herradon B, 2002 Computational studies on biphenyl derivatives. Analysis of the conformational mobility, molecular electrostatic potential, and dipole moment of chlorinated biphenyl: searching for the rationalization of the selective toxicity of polychlorinated biphenyls (PCBs). Chem Res Toxicol 15: 1514-1526.
8. Petersen SL, Curran MA, Marconi SA, Carpenter CD, Lubbers LS, McAbee MD, 2000 Distribution of mRNAs encoding the arylhydrocarbon receptor, arylhydrocarbon receptor nuclear translocator, and arylhydrocarbon receptor nuclear translocator-2 in the rat brain and brainstem. J Comp Neurol 427: 428-439.
9. Seegal RF, Schantz SL 1994 Neurochemical and behavioral sequelae of exposure to dioxins and PCBs. In: Schechter A (ed) Dioxins and Health, Plenum Press; New York; pp, 409-447.
10. Phoenix CH, Goy RW, Gerall AA, Young WC, 1959 Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 65: 369-392.
11. Schwarz JM, McCarthy MM, 2008 Steroid-induced sexual differentiation of the developing brain: multiple pathways, one goal. J Neurochem 105: 1561-1572.
12. Gore AC, 2008 Developmental exposures and imprinting on reproductive neuroendocrine systems. Front Neuroendocrinol 29: 358-374.
13. Barker DJP, 2003 The developmental origins of adult disease. Eur J Epidemiol 18: 733-736.
14. Patisaul HB, Polston EK, 2008 Influence of endocrine active compounds on the developing rodent brain. Brain Res Rev 57: 352-362.
15. Patisaul HB, Luskin JR, Wilson ME, 2004 A soy supplement and tamoxifen inhibit sexual behavior in female rats. Horm Behav 45: 270-277.
16. Patisaul HB, Fortino AE, Polston EK, 2006 Neonatal genistein or bisphenol-A exposure alters sexual differentiation of the AVPV. Neurotoxicol Teratol 28: 111-118.
17. Lephart ED, Setchell KD, Handa RJ, Lund TD, 2004 Behavioral effects of endocrine-disrupting substances: Phytoestrogens. ILAR J 45: 443-454.
18. Simerly RB, 1989 Hormonal control of the development and regulation of tyrosine hydroxylase expression within a sexually dimorphic population of dopaminergic cells in the hypothalamus. Mol Brain Res 6: 297-310.
19. Hutton LA, Gu G, Simerly RB, 1998 Development of a sexually dimorphic projection from the bed nuclei of the stria terminalis to the anteroventral periventricular nucleus in the rat. J Neurosci 18: 3003-3013.
20. Simerly RB, 1998 Organization and regulation of sexually dimorphic neuroendocrine pathways. Behav Brain Res 92: 195-203.
21. Bakker J, Baum MJ, 2008 Role for estradiol in female-typical brain and behavioral sexual differentiation. Front Neuroendocrinol 29: 1-16.
22. Balthazart J, De Clerck A, Foidart A, 1992 Behavioral demasculinization of female quail is induced by estrogens: studies with the new aromatase inhibitor R76713. Horm Behav 26: 179-203.
23. Roselli CE, 1991 Sex differences in androgen receptors and aromatase activity in microdissected regions of the rat brain. Endocrinology 128: 1310-1316.
24. Bakker J, De Mees C, Douhard Q, et al, 2006 Alpha-fetoprotein protects the developing female mouse brain from masculinization and defeminization by estrogens. Nat Neurosci 9: 220-226.
25. Forger NG, 2006 Cell death and sexual differentiation of the nervous system. Neuroscience 138: 929-938.
26. Arai Y, Murakami S, Nishizuka M, 1994 Androgen enhances neuronal degeneration in the developing pre¬optic area: apoptosis in the anteroventral periventricular nucleus (AVPV-POA). Horm Behav 28: 313-319.
27. Davis EC, Popper P, Gorski RA, 1996 The role of apoptosis in sexual differentiation of the rat sexually dimorphic nucleus of the preoptic area. Brain Res 734: 10-18.
28. Patisaul HB, Fortino AE, Polston EK, 2007 Differential disruption of nuclear volume and neuronal phenotype in the preoptic area by neonatal exposure to genistein and bisphenol-A. Neurotoxicol 28: 1-12.
29. Jacobson CD, Shryne JE, Shapiro F, Gorski RA, 1980 Ontogeny of the sexually dimorphic nucleus of the preoptic area. J Comp Neurol 193: 541-548.
30. Simerly RB, 2002 Wired for reproduction: organization and development of sexually dimorphic circuits in the mammalian forebrain. Ann Rev Neurosci 25: 507-536.
31. Chung YW, Nunez AA, Clemens LG, 2001 Effects of neonatal polychlorinated biphenyl exposure on female sexual behavior. Physiol Behav 74: 363-370.
32. Steinberg RM, Juenger TE, Gore AC, 2007 The effects of prenatal PCBs on adult female paced mating reproductive behaviors in rats. Horm Behav 51: 364-372.
33. Laessig SA, Auger AP, McCarthy MM, Silbergeld EK, 2007 Effects of prenatal chlordecone on sexually differentiated behavior in adult rats. Neurotoxicol Teratol 29: 255-263.
34. Whitten PL, Lewis C, Russell E, Naftolin F, 1995 Phytoestrogen influences on the development of behavior and gonadotropin function. Proc Soc Exp Biol Med 208: 82-86.
35. Kouki T, Okamoto M, Wada S, Kishitake M, Yamanouchi K, 2005 Suppressive effect of neonatal treatment with a phytoestrogen, coumestrol, on lordosis and estrous cycle in female rats. Brain Res Bull 64: 449-454.
36. Faass O, Schlumpf M, Reolon S, et al, 2009 Female sexual behavior, estrous cycle and gene expression in sexually dimorphic brain regions after pre- and postnatal exposure to endocrine active UV filters. Neurotoxicol 30: 249-260.
37. Gore AC 2002 GnRH: The Master Molecule of Reproduction. Kluwer Academic Publishers, Norwell, MA.
38. Simerly RB, Chang C, Muramatsu M, Swanson LW, 1990 Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: An in situ hybridization study. J Comp Neurol 294: 76-95.
39. Chakraborty TR, Gore AC, 2004 Aging-related changes in ovarian hormones, their receptors, and neuroendocrine function. Exp Biol Med 229: 977-987.
40. Gore AC, Wu TJ, Oung T, Lee JB, Woller MJ, 2002 A novel mechanism for endocrine-disrupting effects of polychlorinated biphenyls: Direct effects on gonadotropin-releasing hormone (GnRH) neurons. J Neuroendocrinol 14: 814-823.
41. Belsham DD, Evangelou A, Roy D, Le DV, Brown TJ, 1998 Regulation of gonadotropin-releasing hormone (GnRH) gene expression by 5-alpha dihydrotestosterone in GnRH-secreting GT1-7 hypothalamic neurons. Endocrinology 139: 1108-1114.
42. Roy D, Angelini NL, Belsham DD, 1999 Estrogen directly represses gonadotropin-releasing hormone (GnRH) gene expression in estrogen receptor-alpha (ERalpha) and ERbeta-expressing GT1-7 GnRH neurons. Endocrinology 140: 5045-5053.
43. Hrabovszky E, Steinhauser A, Barabás et al, 2001 Estrogen receptor-β immunoreactivity in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology 142: 3261-3264.
44. Wintermantel TM, Campbell RE, Porteous R, et al, 2006 Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52: 271-280.
45. Gore AC, 2002 Organochlorine pesticides directly regulate gonadotropin-releasing hormone gene expression and biosynthesis in the GT1-7 hypothalamic cell line. Mol Cell Endocrinol 192: 157-170.
46. Cook R, Calabrese EJ, 2006 The importance of hormesis to public health. Environ Health Perspect 114: 1631-1635.
47. Dickerson SM, Guevara E, Woller MJ, Gore AC, 2009 Cell death mechanisms in GT1-7 GnRH cells exposed to polychlorinated biphenyls PCB74, PCB118, and PCB153. Toxicol Appl Pharmacol 237: 237-245.
48. Rasier G, Parent AS, Gérard A, et al, 2008 Mechanisms of interaction of endocrine-disrupting chemicals with glutamate-evoked secretion of gonadotropin-releasing hormone. Toxicol Sci 102: 33-41.
49. Rasier G, Parent A, Gérard A, Lebrethon M, Bourguignon J, 2007 Early maturation of gonadotropin-releasing hormone secretion and sexual precocity after exposure of infantile female rats to estradiol or dichlorodiphenyltrichloroethane. Biol Reprod 77: 734-742.
50. Clements RJ, Lawrence RC, Blank JL, 2009 Effects of intrauterine 2,3,7,8-tetrachlorodibenzo-p-dioxin on the development and function of the gonadotrophin releasing hormone neuronal system in the male rat. Reprod Toxicol 28: 38-45.
51. McGarvey C, Cates PA, Brooks A, et al, 2001 Phytoestrogens and gonadotropin-releasing hormone pulse generator activity and pituitary luteinizing hormone release in the rat. Endocrinology 142: 1202-1208.
52. Khan IA, Thomas P, 2001 Disruption of neuroendocrine control of luteinizing hormone secretion by aroclor 1254 involves inhibition of hypothalamic tryptophan hydroxylase activity. Biol Reprod 64: 955-964.
53. Khan IA, Thomas P, 1997 Aroclor 1254-induced alterations in hypothalamic monoamine metabolism in the Atlantic croaker (Micropogonias undulatas): correlation with pituitary gonadotropin release. Neurotoxicology 18: 553-560.
54. Thomas AK, Mander JS, Hale JS, Walstab J, Forrest M, 1989 Induction of ovulation with subcutaneous pulsatile gonadotropin-releasing hormone: Correlation with body weight and other parameters. Fertil Steril 51: 786-790.
55. Bisenius ES, Veeramachaneni DN, Sammonds GE, Tobet S, 2006 Sex differences and the development of the rabbit brain: effects of vinclozolin. Biol Reprod 75: 469-476.
56. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al, 2009 Endocrine-disrupting chemicals: An endocrine society scientific statement. Endocr Rev 30: 293-342.
57. Howdeshell KL, 2002 A model of the development of the brain as a construct of the thyroid system. Environ Health Perspect 110:Suppl 3: 337-348.
58. Langer P, Kocan A, Tajtakova M, Trnovec T, Klimes I, 2009 What we learned from the study of exposed population to PCBs and pesticides. Open Environ Pollution & Toxicol Journal 1: 54-65.
59. Zoeller RT, Dowling AL, Vas AA, 2000 Developmental exposure to polychlorinated biphenyls exerts thyroid hormone-like effects on the expression of RC3/neurogranin and myelin basic protein messenger ribonucleic acids in the developing rat brain. Endocrinology 141: 181-189.
60. Khan MA, Hansen LG, 2003 Ortho-substituted polychlorinated biphenyl (PCB) congeners (95 or 101) decrease pituitary response to thyrotropin releasing hormone. Toxicol Lett 144: 173-182.
61. Kuriyama SN, Wanner A, Fidalgo-Neto AA, Talsness CE, Koerner W, Chahoud I, 2007 Developmental exposure to low-dose PBDE-99: tissue distribution and thyroid hormone levels. Toxicology 242: 80-90.
62. Piedrafita B, Erceg S, Cauli O, Felipo V, 2008 Developmental exposure to polychlorinated biphenyls or methylmercury, but not to its combination, impairs the glutamate-nitric oxide-cyclic GMP pathway and learning in 3-month-old rats. Neuroscience 154: 1408-1416.
63. Nguon K, Baxter MG, Sajdel-Sulkowski EM, 2005 Perinatal exposure to polychlorinated biphenyls differentially affects cerebellar development and motor functions in male and female rat neonates. Cerebellum 4: 112-122.
64. Zoeller RT, Bansal R, Parris C, 2005 Bisphenol-A, an environmental contaminant that acts as a thyroid hormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expression in the developing rat brain. Endocrinology 146: 607-612.
65. Newbold RR, Padilla-Banks E, Jefferson WN, 2006 Adverse effects of the model environmental estrogen diethylstilbestrol (DES) are transmitted to subsequent generations. Endocrinology 147: S11-S17.
66. Grun F, Blumberg B, 2005 Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 147: Suppl: S50-S55.
67. Anway MD, Cupp AS, Uzumcu M, Skinner MK, 2005 Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308: 1466-1469.
68. Steinberg RM, Walker DM, Juenger TE, Woller MJ, Gore AC, 2008 The effects of prenatal PCBs on adult female rat reproduction. Development, reproductive physiology, and transgenerational effects. Biol Reprod 78: 1091-1101.
69. Jirtle RL, Skinner MK, 2007 Environmental epigenomic and disease susceptibility. Nature Rev Genet 8: 253-262.
70. Weaver IC, D’ Alessio AC, Brown SE, et al, 2007 The transcription factor nerve growth factor-inducible protein A mediates epigenetic programming: Altering epigenetic marks by immediate-early genes. J Neurosci 27: 1756-1768.
71. Weaver IC, Champagne FA, Brown SE, et al, 2005 Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci 25: 11045-11054.
72. Champagne FA, Weaver IC, Diorio J, et al, 2006 Maternal care associated with methylation of the estrogen receptor-1b promoter and estrogen receptor- expression in the medial preoptic area of female offspring. Endocrinology 147: 2909-2915.
73. Cameron N, Del Corpo A, Diorio J, McAllister K, Sharma S, Meaney MJ, 2008 Maternal programming of sexual behavior and hypothalamic-pituitary-gonadal function in the female rat. PLoS ONE 3 (e2210): 1-12.
74. Anway MD, Leathers C, Skinner MK, 2006 Endocrine disruptor vinclozolin induced epigenetic transgenerational adult onset disease. Endocrinology 147: 5515-5523.
75. Schneider S, Kaufmann W, Buesen R, van Ravenzwaay B, 2008 Vinclozolin - The lack of a transgenerational effect after oral maternal exposure during organogenesis. Reprod Toxicol 25: 352-360.
76. Inawaka K, Kawabe M, Takahashi S, et al, 2009 Maternal exposure to anti-androgenic compounds, vinclozolin, flutamide and procymidone, has no effects on spermatogenesis and DNA methylation in male rats of subsequent generations. Toxicol Appl Pharmacol 237: 178-187.
77. Stouder C, Paoloni-Giacobino A, 2010 Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction 139: 373-379.
78. Vandegehuchte MB, Lemiere F, Vanhaecke L, Van den Berghe W, Janssen CR, 2009 Direct and transgenerational impact on Daphnia magna of chemicals with a known effect on DNA methylation. Comp Biochem Physiol C Toxicol Pharmacol. [Epub ahead of print].
79. Bayley M, Larsen PF, Baekgaard H, Baatrup E, 2003 The effects of vinclozolin, an anti-androgenic fungicide, on male guppy secondary sex characters and reproductive success. Biol Reprod 69: 1951-1956.
80. Crews D, Gore AC, Hsu TS, et al, 2007 Transgenerational epigenetic imprints on mate preference. Proc Natl Acad Sci USA 104: 5942-5946.
81. Skinner MK, Anway MD, Savenkova MI, Gore AC, Crews D, 2008 Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS ONE 3 (e3745): 1-11.
Address for correspondence:
Andrea C. Gore, Ph.D. The University of Texas at Austin,
Austin, TX 78712, USA, Tel.: 512-471-3669,
Fax: 512-471-5002, Email: andrea.gore@mail.utexas.edu
Received 22-09-09, Revised 10-11-09, Accepted 10-12-09