Endocrine practice, Heidelberg, Germany
Multiple endocrine neoplasia type 2 (MEN2) is an autosomal dominant tumour syndrome caused by germline activating mutations of the RET proto-oncogene. It has a strong penetrance of medullary thyroid carcinoma (MTC) and can be associated with bilateral pheochromocytoma and primary hyperparathyroidism (MEN2A) within a single patient or family. Based on the phenotype three distinct clinical forms have been described: (1) classical MEN2A, (2) MEN2B, an association of MTC, pheochromocytoma and mucosal neuroma and (3) familial MTC (FMTC), which is associated with a very low incidence of other endocrinopathies. Each variant of MEN2 results from a different RET gene mutation, with a good genotype-phenotype correlation with regard to aggressiveness of MTC, time of onset of MTC and the presence or absence of other endocrine tumours. Recommendations on the timing of prophylactic thyroidectomy and extent of surgery are based on a classification of RET mutations into three risk levels using the genotype-phenotype correlations. MEN2 provides a unique model for early prevention and cure of cancer and for stratified roles of mutation-based diagnosis of carriers.
Medullary thyroid carcinoma, Multiple endocrine neoplasia type 2, Pheochromocytoma, Primary hyperparathyroidism, RET gene mutation
Α. INTRODUCTION
Multiple endocrine neoplasia type 2 (MEN2) (OMIM 171400) is an autosomal dominant syndrome with an estimated prevalence of 2.5 per 100,000 in the general population. Predisposition to MEN2 is caused by germline activating mutations of the RET proto-oncogene.1,2 MEN2 shows a high penetrance for medullary thyroid carcinoma (MTC), a rare calcitonin (CT)-secreting tumour of the parafollicular or C-cells of the thyroid. Most patients suffer from the sporadic (non-familial) form, while 25-30% account for hereditary MTC. Hereditary MTC is usually bilateral and multicentric; multifocal C-cell hyperplasia is considered to be a precursor of MTC in patients with hereditary disease. Although MEN2 syndromes are rare, recognition is important both for treatment and for genetic counselling.
CLINICAL SYNDROMES OF MEN2 (Table 1 )
MEN2 syndrome occurs in three clinically distinct varieties with MTC as the most common manifestation. The three subtypes of MEN2 differ with respect to incidence, genetics, age of onset, association with other diseases, aggressiveness of MTC and prognosis.1,2
MEN2A SYNDROME
MEN2A syndrome is the most common form and is characterized by MTC in combination with pheochromocytoma and/or multiple tumours of the parathyroid glands in a single patient, or the presence of two or more tumour types in multiple members of a single family.2 Among patients with MEN2A, the frequency of MTC is over 90%, of pheochromocytoma 40-50%, and of multiple parathyroid gland hyperplasia or a single adenoma 10-20%. MTC is generally the first manifestation of MEN2A and develops between the ages of 5 to 25 years. Some rare variants of MEN2A exist, including MEN2A associated with cutaneous lichen amyloidosis, and FMTC (or MEN2A) with Hirschsprung’s disease.
MEN2B Syndrome
MEN2B syndrome is the least common but the most aggressive form of MEN2 (5-10% of all cases). It consists of MTC, pheochromocytoma, without hyperparathyroidism, and visible physical stigmata like bumpy lips and tongue (neuromas) and a marfanoid habitus. Patients with MEN2B typically experience disease onset in the first year of life and have a more aggressive form of MTC with higher morbidity and mortality rates compared with patients with MEN2A. Patients with MEN2B also have mucosal neuromas, intestinal ganglioneuromas and a marfanoid habitus with skeletal deformations and joint laxity but without the vascular and ophthalmologic abnormalities. They often do not have a family history of the disease and harbour a de novo mutation.
FMTC Syndrome
Familial MTC (FMTC) is the mildest variant of MEN2. It has been diagnosed more frequently in recent years (35-40% of all cases).3-5 In patients with FMTC, there is a strong predisposition to develop MTC with a very low incidence of the other clinical manifestations of MEN2A. The diagnosis of FMTC can only be considered when four or more family members across a wide range of ages have isolated MTC. The clinical course of MTC in FMTC in general is more benign than for individuals with MEN2A and MEN2B and typically has a late onset or no clinically manifest disease. The prognosis for FMTC is relatively good; however, aggressive MTC tumours and even death due to MTC have been reported in cases harbouring codon 804 mutations. A family history is often inadequate in establishing diagnosis of familial disease, a more thorough evaluation by genetic and biochemical screening often revealing a family history of MTC in patients originally thought to have the sporadic form of the disease.
RET PROTO-ONCOGENE: STRUCTURE, FUNCTION AND MUTATIONS
The RET gene has 21 exons and encodes a tyrosine kinase receptor that appears to transduce growth and differentiation signals in several developing tissues including those derived from the neural crest. The protein consists of an extracellular part with a ligand-binding domain, a cadherin (Ca2+-dependent cell adhesion)-like domain and a cysteine-rich domain close to the cell membrane. It has a single transmembrane domain and an intracellular part with two tyrosine kinase subdomains, TK1 and TK2. The RET protein is activated upon ligand-induced dimerization.6 RET is expressed in neuroendocrine cells such as C-cells, the precursors of MTC, and in pheochromocytomas. Hereditary MTC is caused by autosomal dominant gain-of-function mutations in the RET proto-oncogene.
The MEN2 gene was localized to centromeric chromosome 10 (10q11.2) by genetic linkage analysis in 1987. Subsequently, point mutations of the RET proto-oncogene were identified in MEN2A, MEN2B and FMTC in 7 exons located nearby (exons 8, 10, 11, 13-16).7,8 Analysis of RET in families with MEN2A and FMTC revealed that nearly 100% of these families have germline mutations and that only those family members with the germline missense mutation had the disease. This discovery prompted major advances in our understanding of the molecular genetic basis of MTC and has significantly changed the clinical management of families with hereditary tumours. At present, mutation analysis in MEN2 families has identified over 50 different missense mutations segregating with the disease.5
Mutation of the extracellular cysteine at exon 11 codon 634 causes ligand-independent dimerization of receptor molecules, enhanced phosphorylation of intracellular substrates and cell transformation. Mutation of the intracellular tyrosine kinase (codon 918) has no effect on receptor dimerization but causes constitutive activation of intracellular signalling pathways and also results in cellular transformation. There is a significant age-related progression from C-cell hyperplasia (CCH) to MTC, which correlates with the transforming capacity of the respective RET mutations.9 MTC is generally the first neoplastic manifestation because of its earlier and higher penetrance compared with pheochromocytoma or parathyroid hyperplasia. This indicates that C cells are more susceptible to oncogenic RET activation than adrenal medullary or parathyroid cells.
CHANGE IN THE SPECTRUM OF DETECTED RET MUTATIONS OVER THE LAST TWO DECADES
Our analysis of the RET proto-oncogene in patients with hereditary MTC, diagnosed between 1994 and 2006, provided evidence for a change in the spectrum of detected mutations: increase of diagnosed cases from 10% to 39% of the families with mutations in exon 13-15, so called “rare mutations” (Figure 1).5
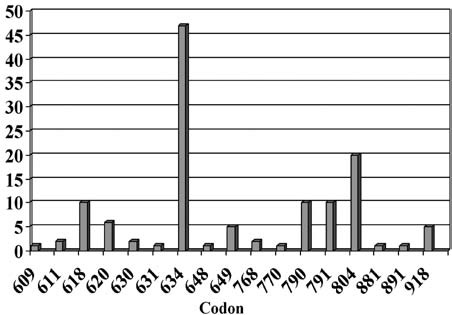
Figure 1. Distribution of different RET mutations (130 families) identified between 2000 and 2008 (Laboratory of Molecular Genetics, endocrine practice, Heidelberg).
This change in the frequency of diagnosed mutations in families with classical MEN2A from mutations mainly in exon 11 codon 634 (formerly 85% of all MEN2A mutations) to the so-called rare mutations localized at codons10 13, 14 and 15 may also have impact on the overall prognosis of hereditary MTC. The reasons for this change in the spectrum may be attributed to the improvement in diagnostics, since RET mutations are routinely looked for in all patients diagnosed with MTC, not only familial but apparently sporadic cases as well (4-7% hereditary). Furthermore, analysis involves not only known hot spots but other known mutations.4 It must be stressed that there is a distinct distribution of RET mutations in different parts of the world depending on the genetic background.
GENOTYPE-PHENOTYPE CORRELATION IN MEN2
Clear associations between specific RET mutations (genotype) and the age at onset and aggressiveness of MTC and the presence or absence of other endocrine neoplasms (phenotype), such as pheochromocytoma or hyperparathyroidism, are well documented.10,11 Although some overlap exists between RET mutations and the resulting clinical subtype of MEN2, 85 % of patients with MEN2A have the codon 634 (exon 11, cystein–rich domain) mutation, mutations of codon 609, 611, 618 and 620 accountings for a further 10% to 15%.
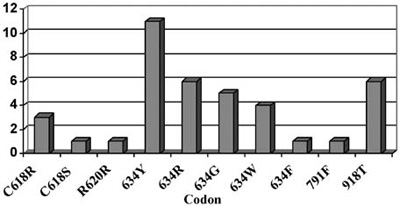
Figure 2. Pheochromocytoma in MEN2. The distribution of patients with pheochromocytoma in the different RET mutations (endocrine practice, Heidelberg) in a series of 130 families with RET mutations.
Pheochromocytomas are also detected in about 50% of patients with 634 and 918 codon mutations and are rarely seen in mutations of exon 10 (codon 609, 611, 618, 620) or exon 15 (codon 791, 804) (Figure 2).12,13 Hyperparathyroidism in MEN2A is most commonly associated with codon 634 mutations, and in particular with the C634R mutation14 (Figure 3).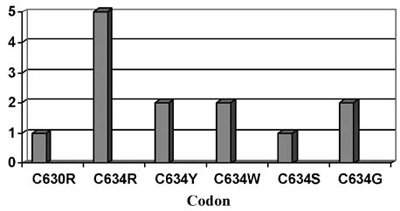
Figure 3. Primary Hyperparathyroidism (HPT) in MEN2. Distribution of patients with HPT in the different RET mutations (endocrine practice, Heidelberg) in a series of 130 families with RET mutations.
The association between disease phenotype and RET mutation genotype may have important implications for the clinical management of MEN2 patients and their families. If the genotype can be fully correlated with certain phenotypic features, then a clinician could use a patient’s genotype to decide whether to intensify screening for pheochromocytoma or hyperparathyroidism for those patients with mutations associated with a higher risk.
All cases of MEN2A with Hirschspung’s disease have mutations in exon 10 (codon 609, 611, 618, 620), while patients with MEN2A and cutaneous lichen amyloidosis are associated with mutations in codon 634.
More than 95% of MEN2B patients have mutations in codon 918 (exon 16, tyrosine kinase domain) and, rarely in codon 883 of exon 15. The 918 mutation results in an ATG (methionine) to ACG (threonine) alteration, which is significant since Met 918 is a critical component of the substrate recognition pocket in the tyrosine kinase catalytic core of the RET protein.
In FMTC, germline mutations are distributed throughout the RET gene with an accumulation in exon 13 (codon 768, 790, 791) and exon 14 (codon 804,844). Some of these mutations have also been identified in families with MEN2A. Because FMTC shares a common genetic defect with MEN2A, it can be difficult to distinguish a family that initially appears to be FMTC from MEN2A, as the manifestation of pheochromocytoma and/or hyperparathyroidism occurs over time.
PATIENT RISK GROUPS
Extensive literature shows a correlation between the specific germline RET mutation and the age of onset and aggressiveness of MTC development and nodal metastases.9 Patients with codon 918 mutation and MEN2B have a high risk for aggressive MTC at young age. In contrast, patients with codon 791 mutations have a relatively low risk of a late manifestation and slowly growing tumour.15 This genotype-phenotype correlation of age at onset and aggressiveness of MTC is the basis for decision making in clinical management, especially in pre-symptomatic RET mutation carriers, as prophylactic thyroidectomy has to be performed prior to the development of cancer. This strategy for prevention of familial MTC should be tailored to the specific mutation carried by the individual patient.
Recommendations for the timing of prophylactic thyroidectomy and the extent of surgery are based upon a model that utilizes these genotype-phenotype correlations to stratify mutations into three risk levels.16,17 Patients with level 1 mutations (codons 609, 768, 790, 791, 804 and 891) have a high risk for MTC development and growth, patients with level 2 mutations (codons 611, 618, 620 and 634) are higher risk patients, and patients with level 3 mutations (codons 883 and 918) are at the highest risk for early development and growth of MTC (Table 2 ).
Codon-specific prognosis would facilitate individual risk stratification for each patient. Furthermore, good evidence exists of a significant age-related progression from C-cell hyperplasia (CCH) to MTC that correlates with the transforming potential of level 2 (codon 634) and level 3 mutations (codon 918).9 In patients with the higher risk mutations (level 2), a thyroidectomy is recommended at the age of five years, and as early as possible (preferably in the first year after birth) for patients with level 3 mutations.16 For patients with level 1 mutations there are three alternatives concerning recommended age at prophylactic surgery: some authors suggest 5, others 10 years of age, others, as the authors do,15 suggest that surgery may be postponed until after an abnormal C cell stimulation test result is observed (i.e. an abnormal CT response to pentagastrin or calcium stimulation). Further studies, especially in the rare mutations,18 are necessary before more specific recommendations can be made (Table 3 ).
In recent years a certain variability in clinical presentation among genetically related individuals has been observed. This intrafamilial phenotypic variability regarding the age at onset and disease extent at diagnosis (tumour size, presence of lymph nodes or distant metastases) is a matter of further investigation in this field. It has been speculated that interacting genetic alterations may be responsible for this phenotypic heterogeneity. Single Nucleotide Polymorphisms (SNPs) within the RET oncogene have been reported (G691S, L769L, S836S, and S904S) and the modifying role of the clinical phenotype of MEN2 have been discussed.19,20 Only the RET G691S variant could modulate the age of onset of familial and sporadic MTC with higher basal calcitonin levels.21 We have confirmed the data of Cardot-Bauters21 in our series of 66 patients with hereditary MTC with regard to the aggressiveness of the MTC; we found that tumour stage increase was related to the frequency of G691S variants in patients with MEN2 (Figure 4).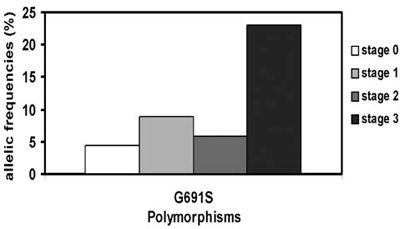
Figure 4. Allelic frequencies of G691S and tumour stage at diagnosis in 56 patients with MEN2.
PERSPECTIVES
The biological behaviour of MTC can now be stratified by mutations of the RET proto-oncogene, which guides the timing of prophylactic thyroidectomy and the extent of surgery. This has to be established for rare mutations in order to refine the codon specific treatment.
Advances in our understanding of the molecular pathways underlying the different phenotypes of MEN2 may help to tailor more individualized therapeutic modalities based on codon-specific inhibition of tumour growth.
REFERENCES
1. Raue F, Frank-Raue K, 2007 Multiple Endocrine Neoplasia Type 2: 2007 Update. Horm Res 68:101-104.
2. Kouvaraki MA, Shapiro SE, Perrier ND, et al, 2005 RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 15: 531-544.
3. Berndt I, Reuter M, Saller B, et al, 1998 A new hotspot for mutations in the RET proto-oncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 83:770–774.
4. Elisei R, Romei C, Cosci B, et al, 2007 RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab 92: 4725-4729.
5. Frank-Raue K, Rondot S, Schulze E, Raue F, 2007 Change in the spectrum of RET mutations diagnosed between 1994 and 2006. Clin Lab 53: 273-282.
6. Hubner RA, Houlston RS, 2006 Molecular advances in medullary thyroid cancer diagnosis. Clin Chim Acta 370: 2-8.
7. Mulligan LM, Kwok JBJ, Healey CS, et al, 1993 Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2a (MEN 2A). Nature 363: 458-469.
8. Donis-Keller H, Dou S, Chi D, et al, 1993 Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 2: 851-856.
9. Machens A, Niccoli-Sire P, Hoegel J, et al, 2003 for the European Multiple Endocrine Neoplasia (EUROMEN) Study Group. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 349:1517-1525.
10. Eng C, Clayton D, Schuffenecker I, et al, 1996 The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276:1575-1579.
11. Yip L, Cote GL, Shapiro SE, et al, 2003 Multiple endocrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg 138: 409-416.
12. Machens A, Brauckhoff M, Holzhausen HJ, Thanh PN, Lehnert H, Dralle H, 2005 Codon-specific development of pheochromocytoma in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab 90: 3999-4003.
13. Quayle FJ, Fialkowski EA, Benveniste R, Moley JF, 2007 Pheochromocytoma penetrance varies by RET mutation in MEN 2A. Surgery 142: 800-805.
14. Schuffenecker I, Virally-Monod M, Brohet R, et al, and le Groupe d’Etude des Tumeurs a Calcitonine 1998 Risk and penetrance of primary hyperparathyroidism in multiple endocrine neoplasia type 2A families with mutations at codon 634 of the RET proto-oncogene. J Clin Endocrinol Metab 83: 487-491.
15. Frank-Raue K, Machens A, Scheuba, C, Niederle B, Dralle H, Raue F, and the German MEN 2 Study Group 2008 Difference in the development of medullary thyroid carcinoma among carriers of RET mutations in codon 790 and 791. Clin Endocrinol 69: 259-263.
16. Brandi ML, Gagel RF, Angeli A, et al, 2001 Guidelines for diagnosis and therapy of MEN Type 1 and Type 2. J Clin Endocrinol Metab 86: 5658-5671.
17. Frank-Raue K, Buhr H, Dralle H, et al, 2006 Long-term outcome in 46 gene carriers of hereditary medullary thyroid carcinoma after prophylactic thyroidectomy: impact of individual RET genotype. Eur J Endocrinol 155: 229-236.
18. Colombo-Benkmann M, Li Z, Riemann B, et al, 2008 Characterization of the RET protooncogene transmembrane domain mutation S649L associated with nonaggressive medullary thyroid carcinoma. Eur J Endocrinol 158:811-816.
19. Robledo M, Gil L, Pollan M, et al, 2003 Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Research 63: 1814-1817.
20. Cebrian A, Lesueur F, Martin S, et al, 2005 Polymorphisms in the initiators of RET (rearranged during transfection. signalling pathway and susceptibility to sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 90: 6268-6274.
21. Cardot-Bauters C, Leteurtre E, Leclerc L, et al, 2008 Does the RET variant G691S influence the features of sporadic medullary thyroid carcinoma? Clin Endocrinol 69: 506-510.
Address for correspondence:
Friedhelm Raue, Endocrine practice Brückenstr. 21, 69120 Heidelberg, Germany, Tel.: +49(0)6221-439090,
Fax: +49(0)6221-439099, e-mail: friedhelm.raue@raue-endokrinologie.de
Received 01-11-08, Revised 25-11-08, Accepted 10-12-08